





A 22-year-old male patient presented with a fever for the previous 10 days, breathlessness with dry cough for the previous 3 days and decreased urine output since the morning of admission. His history was unremarkable. He denied any history of joint pain, rashes, eye symptoms, haemoptysis or sinus disease and there was no history of illicit drug abuse. He was a nonsmoker with no history of alcohol intake and was not on any regular medications.
Clinical examination revealed icterus, blood pressure of 130/85 mmHg, tachycardia (130 beats·min−1), tachypnoea (30 breaths·min−1) and a temperature of 39°C. His upper airways were normal on examination and there were reduced breath sounds and crackles at the right lung base. The rest of the system examinations was unremarkable.
Initial relevant investigations are summarised in table 1. The capillary blood glucose was 117 mg·dL−1. His typhoid and dengue serology along with dual malarial antigens were all-negative. An electrocardiogram showed sinus tachycardia.
Arterial blood gases on air showed type 1 respiratory failure with a pH of 7.39, an oxygen tension (PO2) of 56 mmHg, a carbon dioxide tension (PCO2) of 32 mmHg, bicarbonate 25 mmol·L−1 and lactate of 2.9 mmol·L−1. Urine dipsticks showed 1+ proteinuria.
A chest radiograph was performed (fig. 1).

Chest radiography
Task 1
What does the chest radiograph show?
Answer 1
The chest radiograph shows right and left lower zone patchy consolidation.
A diagnosis of community-acquired pneumonia was made and he was started on high-flow oxygen at 8 L·min−1 with mask, intravenous fluids and empirical antibiotics (penicillin with macrolide) after sending blood cultures for analysis.
On the second day of admission, the patient developed generalised tonic clonic seizures in the early morning and was started on intravenous phenytoin. However, seizures recurred after an initial period of remission and, subsequently, levetiracetam was added.
A computed tomography (CT) scan of the brain and a cerebrospinal fluid (CSF) study was requested. The contrast CT scan of brain showed generalised cerebral oedema with no areas of pathological enhancement and the CSF study showed the following. Total cell count: 20 cells·mm3 (100% lymphocytes); protein: 12 mg·dL−1; sugar: 74 mg·dL−1; and normal adenosine deaminase.
He developed status epilepticus that night in spite of being on two anticonvulsants and needed general anaesthesia with intubation and ventilation to control the seizures. He maintained adequate oxygenation with an inspiratory oxygen fraction (FiO2) of 100% and a positive end-expiratory pressure (PEEP) of 8 cmH2O.
At this point, his inflammatory markers continued to remain elevated, serum calcium and magnesium were 7.6 mg·dL−1 and 2.6 mg·dL−1 respectively. Viral serology results were all negative.
Secretions aspirated through endotracheal tube were sent for microbiology culture tests and showed Pseudomonas sp. An electroencephalogram showed generalised dysrhythmia.
He continued to be dependent on general anaesthesia for seizure control and needed high FiO2 (>80%) to maintain adequate arterial oxygen tension (PaO2).
The serum phenytoin levels were normal 9.84 mg·mL−1 (acceptable therapeutic range: 10–12 mg·mL−1).
A repeat chest radiograph was performed (fig. 2).

Repeat chest radiography
Task 2
What does the chest radiograph show?
Answer 2
The chest radiograph shows that there was almost complete resolution of the right and left basal infiltrates and appearance of new infiltrates in the left upper lobe.
This is a classical picture of migratory pulmonary infiltrates. Infective consolidations are unlikely to resolve completely in 48 h. The differential diagnosis would include:
-
Eosinophilic pneumonia
-
Organising pneumonia
-
Vasculitis
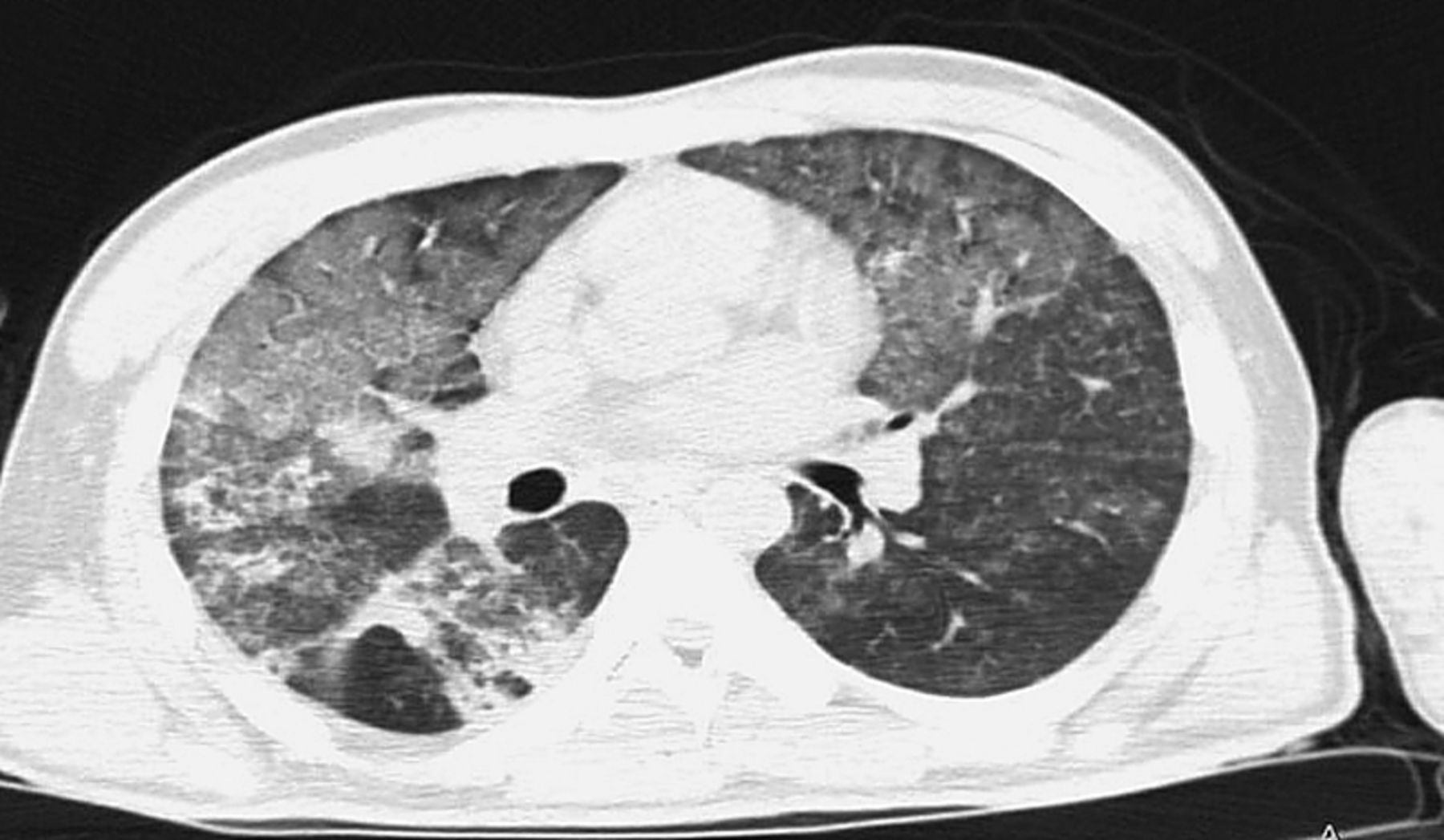
A high-resolution CT scan (HRCT) of chest was performed (fig. 3).
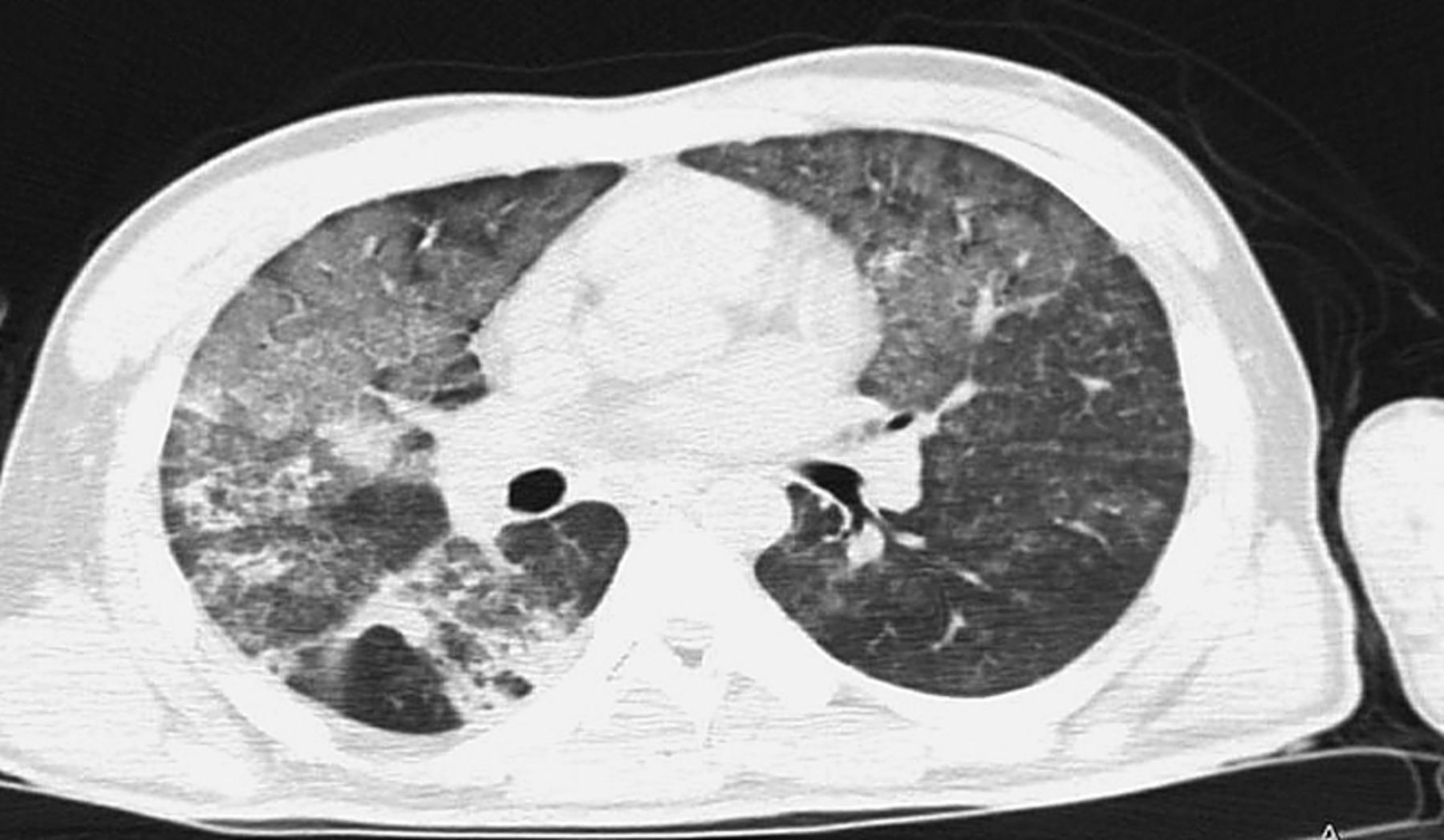
High resolution chest radiography
Task 3
Interpret the HRCT chest scan.
Answer 3
The HRCT show bilateral widespread ground glass opacities with patchy consolidations predominantly peripheral.
The differential diagnosis is shown in table 2.
Echocardiography showed normal left and right ventricular functions, and the serum N-terminal pro-brain natriuretic peptide (NT-pro-BNP) was normal.
Blood for immune markers showed anti-nuclear antibody, rheumatoid factor, complement 3, 4, cytoplasmic and perinuclear anti-neutrophil cytoplasmic antibodies (c-ANCA and p-ANCA), anti-dsDNA antibodies, anti-glomerular basement membrane antibodies, and anti-Jo-1 antibodies were all negative. Cryoglobulins were not detected.
Bronchoscopy with bronchioalveolar lavage (BAL) showed no endobronchial pathology or stenosis. The lavage was not blood stained and serial aspirated aliquots did not produce a consistently bloody return; no haemosiderin-laden macrophages were identified. This would exclude a significant diffuse alveolar haemorrhage. No eosinophils were identified in the BAL fluid and Pneumocystis staining was negative. Culture of the lavage fluid showed growth of Klebsiella sp. and his antibiotics were changed according to the sensitivity.
A transbronchial lung biopsy (TBLB) was also performed.
On the fourth day after admission, he continued to have protracted seizures and remained ventilated. Inflammatory markers remained elevated and there was no improvement in the renal functions. Chest radiograph showed complete resolution of the left upper lobe infiltrates and reappearance of right lower lobe infiltrates (fig. 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Chest radiography showing resolution of left upper lobe infiltrates but reappearance of right lower lobe infiltrates
Task 4
What is the diagnosis?
Answer 4
Small vessel vasculitis (SVV) of the lungs
With persistently raised inflammatory parameters, migratory shadows on chest radiograph, multisystem involvement and inadequate response to appropriate antibiotics, systemic vasculitis was considered and he was started at this stage on intravenous methyl prednisolone, 1 g along with antibiotics while waiting on a biopsy report.
Two days later, there was significant improvement in his clinical status, he was conscious and orientated, maintaining all vital parameters, his inflammatory parameters normalised, renal function test (creatinine 0.9 mg·dL−1, urea 38 mg·dL−1) and liver function tests became normal. There was complete resolution of the pulmonary infiltrates from the chest radiograph 4 days later and repeat HRCT also showed almost complete resolution of the ground glass opacities and consolidation.
His lung biopsy report was available and the final diagnosis was confirmed. The TBLB specimens show vessel walls infiltrated by lymphocytes and neutrophils with areas of fibrin thrombi. This would be consistent with pulmonary capillaritis. There was no infiltration by eosinophils and no evidence of necrosis/granuloma. No evidence of bland alveolar haemorrhage was identified.
Discussion
The pulmonary vasculitides are a rare group of heterogeneous disorders characterised by inflammation of the vessel wall often with associated necrosis. The incidence of ANCA-associated vasculitis (AAV) is only 15–20 cases per million per year [1]. Manifestations of vasculitis can be extremely variable and nonspecific, and can often mimic other common disorders due to overlapping signs and symptoms. Vasculitis is a clinical diagnosis and final diagnosis can only be reached after integration of clinical, laboratory, radiographic, and histopathological data, and making a determination that the preponderance of the data supports or does not support a diagnosis of vasculitis.
Pulmonary involvement in the form of SVV can occur in the setting of systemic vasculitis, collagen vascular disease or any autoantibody-associated systemic illness (table 2) [2]. Isolated pulmonary involvement can also be present, referred to as pauci-immune isolated pulmonary capillaritis (PIPC). This isolated form of SVV of the lungs is one of the most frequent causes of pulmonary capillaritis and diffuse alveolar haemorrhage (DAH). Serum ANCA is usually negative but several p-ANCA-positive cases have been reported [3–6].
SVV of the lungs (capillaritis, arteriolitis and venulitis) and DAH may be the initial and only pulmonary manifestation of granulomatosis with polyangiitis (GPA), previously termed as Wegener's granulomatosis [2].
In GPA, there is usually renal involvement in the form of focal segmental necrotising glomerulonephritis. The combination of these, along with serum c-ANCA can establish the diagnosis.
75–90% of patients of GPA with active disease have antibodies directed against proteinase 3 (PR-3) that produce a diffuse cytoplasmic staining by indirect immunofluorescence (c-ANCA). Antibodies directed against myeloperoxidase (MPO) occur in 5–20% of GPA patients and produce a perinuclear staining by indirect immunofluorescence (p-ANCA). Certain patients with GPA may also be negative for ANCA [7].
The sensitivity and specificity for c-ANCA and PR-3 antibodies in GPA are 85–90% and 95% respectively for generalised active disease [8, 9].
Patients with limited disease or who are in disease remission may still be ANCA positive, but at significantly lower rates (60 and 40%, respectively) [10].
It is to be noted that p-ANCAs can be associated with microscopic polyangiitis (MPA) and Churg-Strauss syndrome (CSS) and idiopathic pauci-immune rapidly progressive glomerulonephritis, but specificity is lower than c-ANCA, as other autoimmune disorders like rheumatoid arthritis (RA) and Goodpasture's syndrome can cause a non-specific rise in p-ANCA titer. p-ANCA/MPO positivity has a sensitivity of 50–75% for MPA and 35–50% for CSS [9, 11–16].
Alternatively, negative ANCA assays do not exclude AAV because between 10% of 50% of patients with ANCA-associated vasculitis (depending on the particular disease) may be ANCA negative [17].
Pulmonary small vessel vasculitis can occur in 10–30% cases of MPA leading to DAH; however, renal involvement is almost universal generally in the form of rapidly progressive glomerulonephritis (80–100%) [2]. GPA without the typical features can be initially misdiagnosed as MPA where the serum p-ANCA is positive [7].
Pulmonary vasculitis in autoimmune diseases
Systemic lupus erythematosus can present with DAH only (23%) where bland pulmonary haemorrhage is found without evidence of vasculitis; more frequently, SVV occurs in association with DAH where immune complex deposition is often found in the small vessels of the lungs [18–21].
Patients with polymyositis can present with muscle disease and SVV; serum ANCA are negative but anti-Jo-1 antibody can be rarely positive [22].
DAH and SVV of the lungs can be a feature of rheumatoid arthritis and mixed connective tissue disease, which can be localised to the lung or be present as a component of a more generalised vasculitis, which often includes glomerulonephritis. Serum p-ANCA levels may be positive [23–25].
Pulmonary capillaritis can complicate the course of scleroderma and primary antiphospholipid syndrome. In the latter, there is usually a history of prior thrombotic events and thrombocytopenia is present [26–29].
This case shows clinical, radiological and pathological evidence of small vessel vasculitis involving the lungs. There was no confirmatory evidence of DAH. There were clinical and laboratory evidence of involvement of kidney and central nervous system which improved and normalised with institution of specific treatment for vasculitis.
Serum ANCA readings were negative and therefore, our case should be categorised into ANCA-negative small vessel vasculitis. It is worth mentioning that ANCA can be negative in a certain percentage of the AAV as discussed above. There was no significant glomerular disease as the urine sediment was not active (no renal biopsy was performed). Autoantibodies for other connective tissue diseases were negative as well. Other conditions leading to small vessel vasculitis (table 3) were been excluded by appropriate investigations. The fact that cryoglobulin levels were normal along with absence of cutaneous and peripheral nerve involvement by the vasculitic process would make the diagnosis of cryoglobulinaemia unlikely [30]. In addition, IgA vasculitis (Henoch–Schonlein disease), which can be one of the conditions leading to pulmonary SVV and DAH, was excluded by the absence of arthritis, skin and gastrointestinal tract involvement [30].
One of our main tools of diagnosis was the histopathology of lungs from the TBLB sample. Because of the small sample size, TBLB can fail to show the features of mild-to-moderate GPA [31]. We do not think this would be relevant in our case as pulmonary involvement was extensive in our case and therefore the TBLB (obtained from an area with significant radiological involvement) was very likely a true representation of the disease.
It can be argued whether our case could be one of a PIPC; because no renal biopsy was done, it would be difficult to establish that there was no glomerulonephritis; however, as mentioned before, urine sediment was inactive without any casts or red blood cells, supporting the fact that kidneys might have been spared. Therefore, we have used the term ANCA-negative small vessel vasculitis of the lungs.
There have been few interesting aspects to the presented case. First, there was no clinical evidence of DAH as shown by almost normal haemoglobin levels and normal bronchoscopy with no evidence of blood stained lavage fluid. This is contrary to published literature, which shows that DAH is almost universal in PIPC.
Secondly, this case initially presented like sepsis and, although there definitely was an element of sepsis, that was not the predominant pathology because the patient failed to improve even after initiation of appropriate antibiotics. Although it can be argued that the renal and hepatic impairment were components of his sepsis, the parameters improved only after starting treatment for vasculitis.
Finally, seizures suggested central nervous involvement by the vasculitic process which has never been reported with isolated pulmonary capillaritis; involvement is more common with other small vessel vasculitis esp. MPA and collagen vascular diseases [32–34].
Conclusion
The diagnosis of pulmonary vasculitis should always be considered in any patient presenting with bilateral pulmonary infiltrates and seizures, which can be intractable unless specific therapy (systemic steroids) are initiated.
Footnotes
Statement of Interest
None declared.
- ©ERS 2014
Breathe articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










