





Abstract
Educational aims
-
To discuss the similarities and differences in inflammation between COPD and asthma
-
To consider the clinical relevance of these differences
-
To speculate about the therapeutic implications of this basic research
Introduction
Asthma and chronic obstructive pulmonary disease (COPD) are both characterised by airway obstruction, which is variable and reversible in asthma but is progressive and largely irreversible in COPD. In both diseases, there is chronic inflammation of the respiratory tract, which is mediated by the increased expression of multiple inflammatory proteins, including cytokines, chemokines, adhesion molecules, inflammatory enzymes and receptors. In both diseases there are acute episodes or exacerbations, when the intensity of this inflammation increases. The similarity between these airway diseases prompted the suggestion in the 1960s that asthma and COPD are part of a spectrum of a common disease (chronic obstructive lung disease) and this came to be known as the “Dutch hypothesis”. This was countered by the “British hypothesis”, which maintained that these diseases were separate entities; the debate continues today, with evidence both for and against these two views [1, 2].
Despite the similarity of some clinical features of asthma and COPD, there are marked differences in the pattern of inflammation that occurs in the respiratory tract, with different inflammatory cells recruited, different mediators produced, distinct consequences of inflammation and differing responses to therapy. In addition, the inflammation seen in asthma is mainly located in the larger conducting airways, although small airways may also be involved in more severe disease, but the lung parenchyma is not affected. By contrast, COPD predominantly affects the small airways and lung parenchyma, although similar inflammatory changes may also be found in larger airways [3, 4]. These differences in disease distribution may partly reflect the distribution of inhaled inciting agents, such as allergens in asthma and tobacco smoke in COPD. In both diseases, there are different recognised clinical phenotypes. Most patients with asthma are atopic (extrinsic asthma), but a few patients are non-atopic (intrinsic asthma) and these patients often have more severe disease [5]. There is a spectrum of asthma severity, which tends to be maintained throughout life [6]. Approximately 5% of patients have severe asthma that is difficult to control with maximal inhaler therapy and for whom new therapeutic approaches are needed. The main types of COPD are the development of small airway obstruction and emphysema, which can occur alone or together, but both involve progressive airflow limitation and are usually caused by tobacco smoke.
The differences in inflammation between asthma and COPD are linked to differences in the immunological mechanisms of these two diseases (figs 1 and 2). There have been several recent important advances in our understanding of the immunopathology of asthma and COPD [7]. T-cells play a crucial role in both asthma and COPD and it is now recognised that different subsets are involved in orchestrating inflammation in these two diseases, resulting in different inflammatory and structural consequences. B-cells also play an important role, although this remains poorly understood in COPD. The appreciation that similar immune mechanisms are involved in both asthma and COPD has important implications for the development of new therapies for these troublesome diseases.

Inflammatory and immune cells involved in asthma. Inhaled allergens activate sensitized mast cells by crosslinking surface-bound immunoglobulin (Ig)E molecules to release several bronchoconstrictor mediators, including cysteinyl-leukotrienes (cys-LT) and prostaglandin D2 (PGD2). Epithelial cells release stem-cell factor (SCF), which is important for maintaining mucosal mast cells at the airway surface. Allergens are processed by myeloid dendritic cells, which are conditioned by thymic stromal lymphopoietin (TSLP) secreted by epithelial cells and mast cells to release the chemokines CC- 31 chemokine ligand (CCL)17 and CCL22, which act on CC-chemokine receptor 4 (CCR4) to attract T helper 2 (Th-2) cells. Th-2 cells have a central role in orchestrating the inflammatory response in allergy through the release of interleukin-4 (IL-4) and IL-13 (which stimulate B-cells to synthesise IgE) IL-5 (which is necessary for eosinophilic inflammation) and IL-9 (which stimulates mast-cell proliferation). Epithelial cells release CCL11, which recruits eosinophils via CCR3. Patients with asthma may have a defect in regulatory T-cells (Treg), which may favour further Th2-cell proliferation.

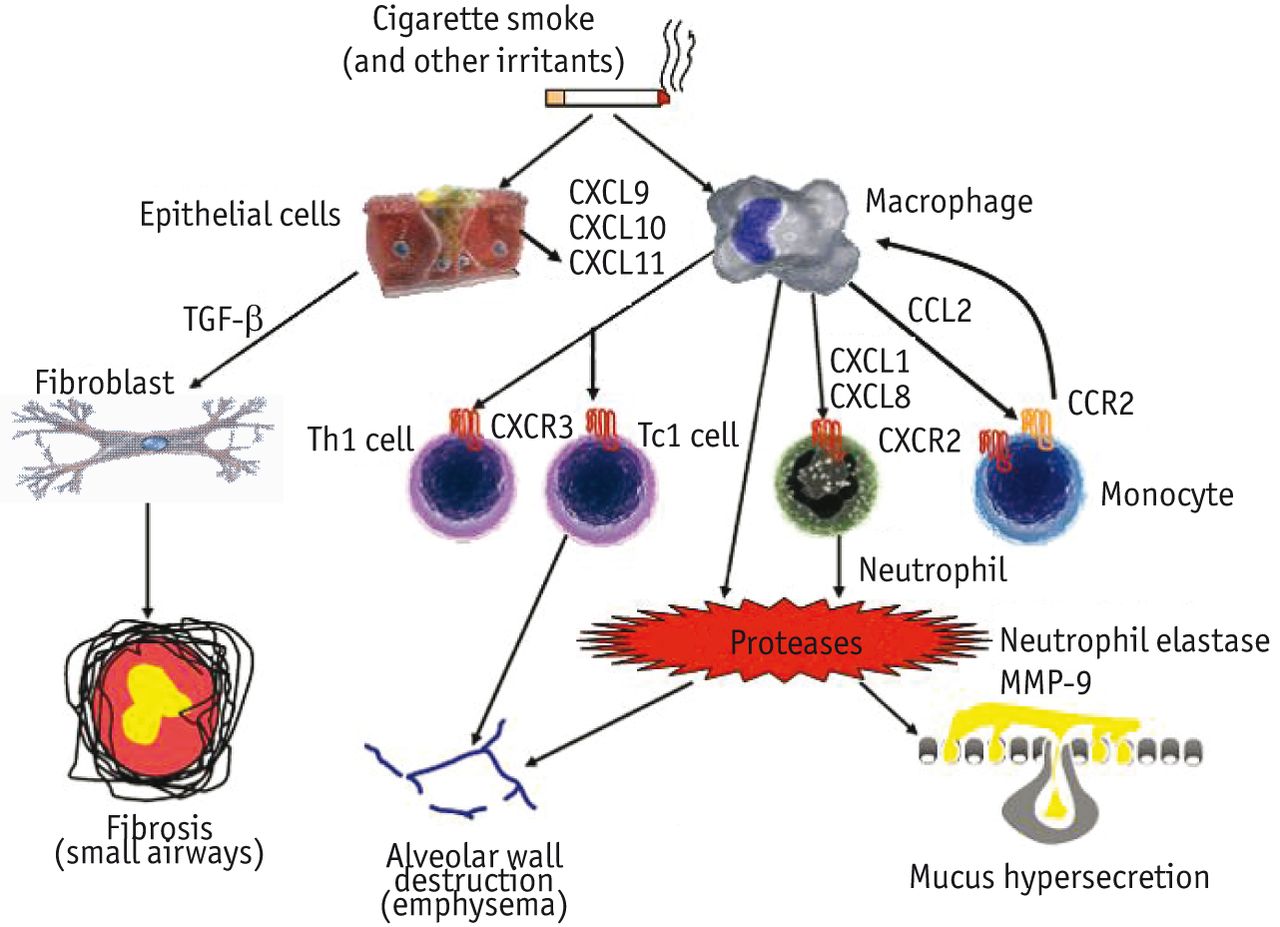
Inflammatory and immune cells involved in chronic obstructive pulmonary disease (COPD). Inhaled cigarette smoke and other irritants activate epithelial cells and macrophages to release several chemotactic factors that attract inflammatory cells to the lungs, including CC-chemokine ligand 2 (CCL2), which acts on CC-chemokine receptor 2 (CCR2) to attract monocytes, CXC-chemokine ligand 1 (CXCL1) and CXCL8, which act on CCR2 to attract neutrophils and monocytes (which differentiate into macrophages in the lungs) and CXCL9, CXCL10 and CXCL11, which act on CXCR3 to attract T helper 1 (Th1) cells and type 1 cytotoxic T-cells (TC1 cells). These inflammatory cells together with macrophages and epithelial cells release proteases, such as matrix metalloproteinase 9 (MMP9), which cause elastin degradation and emphysema. Neutrophil elastase also causes mucus hypersecretion. Epithelia cells and macrophages also release transforming growth factor (TGF), which stimulates fibroblast proliferation, resulting in fibrosis in the small airways.
Inflammatory cells and mediators
There are many differences between mild asthma and COPD in the type of inflammation that occurs in the lungs, with a different range of inflammatory cells and mediators being implicated [8, 9]. However, many of the cytokines and chemokines that are secreted in both asthma and COPD are regulated by the transcription factor nuclear factor (NF)-κB, which is activated in airway epithelial cells and macrophages in both diseases and may have an important role in amplifying airway inflammation.
Histopathology
The histological appearance of airways from asthmatic individuals is very different from the changes that are found in patients with COPD (table 1, fig. 3). Bronchial biopsies from asthmatic subjects reveal an infiltration of eosinophils, activated mucosal mast cells at the airway surface and activated T-cells. There are characteristic structural changes with collagen deposition under the epithelium, which is sometimes described as basement-membrane thickening and is found in all patients, and thickening of the airway smooth-muscle layer as a result of hyperplasia and hypertrophy, which is more commonly seen in patients with severe asthma. Epithelial cells are often shed from patient biopsies compared with normal control biopsies, as they are friable and more easily detach from the basement membrane during the biopsy procedure. In addition, there is an increase in the number of blood vessels (angiogenesis) in response to increased secretion of vascular-endothelial growth factor (VEGF) [10]. Mucus hyperplasia is commonly seen in biopsies from asthmatic patients, with an increase in the number of mucus-secreting goblet cells in the epithelium and an increase in the size of submucosal glands [11].
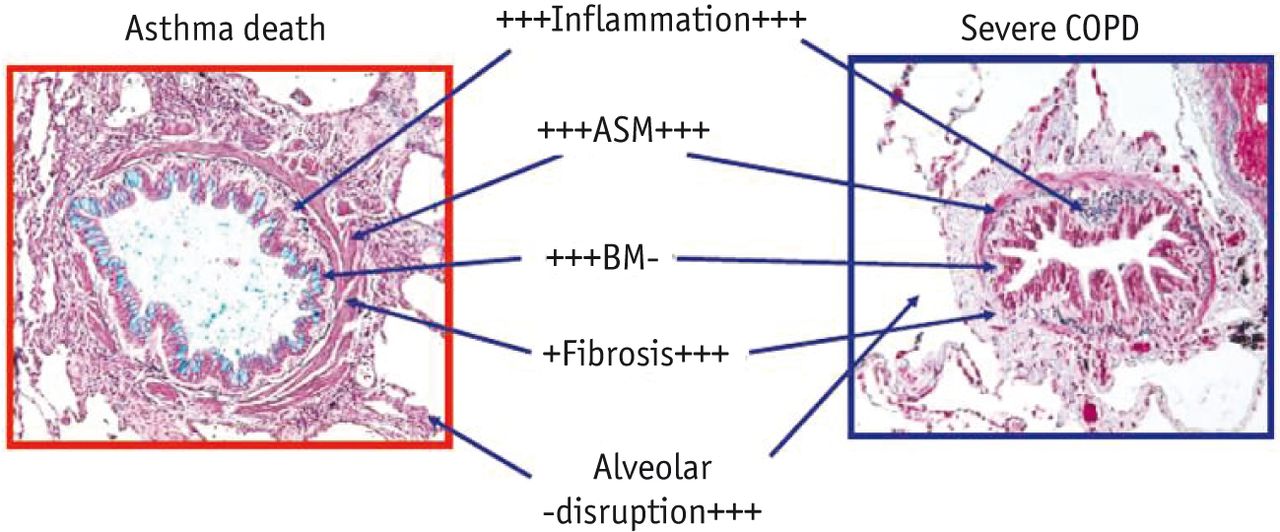
Contrasting histopathology of asthma and chronic obstructive pulmonary disease (COPD). A small airway from a patient who died from asthma and a similar sized airway from a patient with severe COPD are shown. There is an infiltration with inflammatory cells in both diseases. The airway smooth muscle (ASM) layer is thickened in asthma but only to a minimal degree in COPD. The basement membrane (BM) is thickened in asthma due to collagen deposition (subepithelial fibrosis) but not in COPD, whereas in COPD collagen is deposited mainly around the airway (peribronchiolar fibrosis). The alveolar attachments are intact in asthma, but disrupted in COPD as a result of emphysema. Images courtesy of J. Hogg (Vancouver, Canada).
In bronchial biopsies, small airways and lung parenchyma from patients with COPD, there is no evidence for mast-cell activation, but there is an infiltration of T-cells and increased numbers of neutrophils, particularly in the airway lumen [12]. Sub-epithelial fibrosis is not apparent but fibrosis does occur around small airways and is thought to be a major factor that contributes to the irreversible airway narrowing that is characteristic of this disease [13]. Airway smooth muscle is not usually increased in COPD patients compared with normal airways, and airway epithelial cells may show pseudostratification as a result of chronic irritation from inhaled cigarette smoke or other irritants and the release of epithelial-cell growth factors. As in asthma, there is mucus hyperplasia and increased expression of mucin genes in biopsies from patients with COPD [14]. A marked difference between COPD and asthma is the destruction of alveolar walls (emphysema) that occurs in COPD as a result of protease-mediated degradation of connective tissue elements, particularly elastin, as well as apoptosis of type-I pneumocytes and possibly endothelial cells [15, 16]. In addition, the production of elastolytic enzymes, such as neutrophil elastase and particularly several matrix metalloproteinases (MMPs), is increased in the lungs of COPD patients [17], and there may be a reduction in the levels of antiproteinases, such as α1-antitrypsin, as seen in a rare form of emphysema cause by a congenital deficiency of α1-antitrypsin [18].
Mast cells
Mast cells play a key role in asthma through the release of several bronchoconstrictors, including histamine, which is preformed and stored in granules, as well as the lipid mediators leukotriene C4, D4 and E4 and prostaglandin D2, which are synthesised upon mast-cell activation. The release of these mediators may account for the variable bronchoconstriction seen in asthma, as these mediators are released by various environmental triggers, such as allergens, and an increase in osmolality as a result of increased ventilation during exercise. Mucosal mast cells are recruited to the surface of the airways by stem-cell factor (SCF) released from epithelial cells, which acts on KIT receptors expressed by the mast cells [19]. Mast cells also release cytokines that are linked to allergic inflammation, including interleukin (IL)-4, IL-5 and IL-13 [20]. The presence of mast cells in the airway smooth muscle has been linked to airway hyperresponsiveness in asthma [21], as patients with eosinophilic bronchitis have a similar degree of eosinophilic inflammation to that found in asthmatics and also have subepithelial fibrosis, but they do not show hyperresponsiveness, which is the physiological hallmark of asthma. By contrast, mast cells do not seem to play a role in COPD, which may explain the lack of variable bronchoconstriction in this disease.
Granulocytes
The inflammation that occurs in asthma is often described as eosinophilic, whereas that occurring in COPD is described as neutrophilic. These differences reflect the secretion of different chemotactic factors in these diseases. In asthma, eosinophil chemotactic factors, such as CC-chemokine ligand (CCL11, also known as eotaxin-1) [11] and related CC-chemokines, are mainly secreted by airway epithelial cells. The functional role of eosinophils in asthma is not clear and the evidence that links their presence to airway hyperresponsiveness has been questioned by the finding that administration of IL-5-specific blocking antibodies that markedly reduce the number of eosinophils in the blood and sputum does not reduce airway hyperresponsiveness or asthma symptoms [22, 23]. As discussed previously, eosinophilic bronchitis is not associated with airway hyperresponsiveness, but subepithelial fibrosis does occur, which suggests a role for eosinophils in airway fibrosis. Interestingly, the presence of eosinophils seems to be a good marker of steroid responsiveness [24]. Neutrophils are increased in the sputum of patients with COPD and this correlates with disease severity [25]. The increase in neutrophils is related to an increase in the production of CXC-chemokines, such as CXC-chemokine ligand 1 (CXCL1, also known as GROα) and CXCL8 (also known as IL-8), which act on CXCR2 expressed predominantly on neutrophils.
Macrophages
Macrophage numbers are increased in the lungs of patients with asthma and COPD, but their numbers are far greater in COPD than in asthma. These macrophages are derived from circulating monocytes, which migrate to the lungs in response to chemoattractants such as CCL2 (also known as monocyte chemoattractant protein (MCP) 1) acting on CCR2 and CXCL1 acting on CXCR2 [26]. There is increasing evidence that lung macrophages orchestrate the inflammation of COPD through the release of chemokines that attract neutrophils, monocytes and T-cells, and the release of proteases, particularly MMP9 [27]. The pattern of inflammatory cells found in the respiratory tract therefore differs in patients with asthma and those with COPD and some of these contrasts may be explained by differences in the immunological mechanisms driving these two diseases.
Immune responses
The immune mechanisms that drive the different inflammatory processes of asthma and COPD are mediated by different types of immune cell, in particular by different T-cell subsets. An understanding of which immune cells are involved is now emerging and may lead to the development of new and more specific therapies for airway diseases in the future (figs 1 and 2).
T-cells
In asthmatic patients, there is an increase in the number of CD4+ T-cells in the airways and these are predominantly T helper (Th) type 2 cells, whereas, in normal airways Th-1 cells predominate [28]. By secreting the cytokines IL-4 and IL-13, which drive immunoglobulin (Ig)E production by B-cells, IL-5, which is solely responsible for eosinophil differentiation in the bone marrow, and IL-9, which attracts and drives the differentiation of mast cells [29], Th-2 cells have a central role in allergic inflammation and therefore their regulation is an area of intense research.
The transcription factor GATA3 (GATA-binding protein 3) is crucial for the differentiation of uncommitted naïve T-cells into Th-2 cells and it also regulates the secretion of Th-2-type cytokines [30, 31]. Accordingly, there is an increase in the number of GATA3+ T-cells in the airways of asthmatic subjects compared with normal subjects [32, 33]. Following simultaneous ligation of the T-cell receptor (TCR) and co-receptor, CD28, by antigen-presenting cells, GATA3 is phosphorylated and activated by p38 mitogen-activated protein kinase (MAPK). Activated GATA3 then translocates from the cytoplasm to the nucleus, where it activates gene transcription [34]. GATA3 expression in T-cells is regulated by the transcription factor signal transducer and activator of transcription (STAT)6 via IL-4 receptor activation. For Th-1-cell differentiation and secretion of the Th-1-type cytokine, interferon (IFN)-γ, the crucial transcription factor is T-bet. Consistent with the prominent role of Th-2 cells in asthma, T-bet expression is reduced in T-cells from the airways of asthmatic patients compared with non-asthmatic patients [35]. When phosphorylated, T-bet can associate with and inhibit the function of GATA3, by preventing it from binding to its DNA target sequences [36]. T-bet-deficient mice show increased expression of GATA3 and production of Th-2-type cytokines, confirming that T-bet is an important regulator of GATA3 [35]. GATA3 expression is also regulated by IL-27, a recently identified member of the IL-12 family, which downregulates GATA3 expression and upregulates T-bet expression, thereby favouring the production of Th-1-type cytokines, which then act to further inhibit GATA3 expression [37]. In turn, GATA3 inhibits the production of Th-1-type cytokines by inhibiting STAT4, the major transcription factor activated by the T-bet-inducing cytokine IL-12 [38]. Nuclear factor of activated T-cells (NFAT) is a T-cell-specific transcription factor and appears to enhance the transcriptional activation of GATA3 at the IL-4 promoter [39]. Finally, IL-33, a newly discovered member of the IL-1 family of cytokines, seems to promote Th-2-cell differentiation by translocating to the nucleus and regulating transcription through an effect on chromatin structure [40], but it also acts as a selective chemoattractant of Th-2 cells by binding the surface receptor IL-1 receptor-like 1 (also known as ST2), which is specifically expressed by these cells [41].
In contrast to asthma, the CD4+ T-cells that accumulate the airways and lungs of patients with COPD are mainly Th-1 cells. Th-1 cells express the chemokine receptor CXCR3 [42] and may be attracted to the lungs by the IFN-γ-induced release of the CXCR3 ligands CXCL9 (also known as MIG), CXCL10 (also known as IP-10) and CXCL12 (also known as I-TAC), which are present at high levels in COPD airways [42, 43]. However, there is some evidence that Th-2 cells are also increased in lavage fluid of patients with COPD [44]; likewise, in patients with more severe asthma Th-1 cells are activated, as well as Th-2 cells [45], making the distinction between the Th-cell patterns in these two diseases less clear.
Other subtypes of CD4+ T-cells that may play an important role in airway diseases are regulatory T-cells, which have a suppressive effect on other CD4+ T-cells and may play a role in regulating Th-2-cell function in asthma [28, 46]. There is evidence that the numbers of CD4+CD25+ regulatory T-cells that express the transcription factor forkhead box P3 (FOXP3) are reduced in individuals with allergic rhinitis (hay fever) compared with non-atopic individuals, and this may be important in enabling high numbers of Th-2 cells to develop in allergic disease [47]. However, by contrast, asthmatic patients seem to have an increase in FOXP3-expressing regulatory T-cells compared with patients with mild asthma, at least among circulating cells [48]. Analysis of sputum from COPD patients suggests that the numbers of CD4+CD25+FOXP3+ regulatory T-cells are reduced, but similar changes are also seen in people who smoke that do not have airflow obstruction [49]. So, the role of regulatory T-cells in asthma and COPD remains unclear and further research is therefore needed, particularly in defining the role of different types of regulatory T-cells [50].
Another subset of CD4+ T-cells, known as Th-17 cells, has recently been described and shown to have an important role in inflammatory and autoimmune diseases [51, 52]. Little is known about the role of Th-17 cells in asthma or COPD, but increased concentrations of IL-17 (the predominant product of Th-17 cells) have been reported in the sputum of asthma patients [53]. IL-17A and the closely related cytokine IL-17F have been linked to neutrophilic inflammation by inducing the release of CXCL1 and CXCL8 from airway epithelial cells [54]. As well as IL-17, Th-17 cells also produce IL-21, which is important for the differentiation of these cells and thus acts as a positive autoregulatory mechanism, but it also inhibits FOXP3 expression and regulatory T-cell development [55, 56]. Another cytokine, IL-22, is also released by these cells and stimulates the production of IL-10 and acute-phase proteins [57].
However, more work is needed to understand the role and regulation of Th-17 cells in asthma and COPD, as they may represent important new targets for future therapies. A subset of CD4+ T-cells termed invariant natural killer T (iNKT) cells, which secrete IL-4 and IL-13, has been shown to account for 60% of all CD4+ T-cells in bronchial biopsies from asthmatic patients [58], but this has been disputed in another study, which failed to show any increase in iNKT-cell numbers in bronchial biopsies, bronchoalveolar lavage or sputum of either asthma or COPD patients [59]. The role of iNKT-cells in asthma is currently uncertain, as there appears to be a discrepancy between the data from murine models of asthma and humans with the disease [28].
CD8+ T-cells predominate over CD4+ T-cells in the airways and lung parenchyma of patients with COPD [60], but their role in disease pathogenesis is not yet certain. Type 1 cytotoxic T (TC1) cells, which secrete IFN-γ, predominate and express CXCR3, suggesting that they are attracted to the lungs by CXCR3-binding chemokines [44, 61]. These CXCR3 ligands suppress CCR3, the receptor for CCL11, suggesting that they might suppress eosinophilic inflammation [62]. The production of CCL5 (also known as RANTES), which attracts CD4+ and CD8+ T-cells via CCR5, is also increased in the sputum of COPD patients compared with controls and may also be involved in T-cell recruitment [44]. TC1 cells release granzyme B and perforins, which are also present at higher levels in the sputum of COPD patients than in normal control subjects who also smoke [63], and may induce apoptosis of type 1 pneumocytes, thereby contributing to the development of emphysema [15]. TC1- and Th-1-cell-driven inflammation is likely to be self-perpetuating as IFN-γ stimulates the release of CXCR3 ligands, which then attract more Th-1 and TC1 cells into the lungs (fig. 4). TC2 cells, which secrete IL-4, have also been described in COPD [45]. In asthma, CD8+ T-cells are present in patients with more severe disease and irreversible airflow obstruction [64] and these cells may be of either the TC1 or TC2 type [65].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD8+ T-cells in chronic obstructive pulmonary disease (COPD). Epithelial cells and macrophages are stimulated by interferon-γ (IFN) to release the chemokines CXC-chemokine ligand 9 (CXCL9), CXCL10 and CXCL11, which together act on CXC-chemokine receptor 3 (CXCR3) expressed on T helper 1 (Th1) cells and type 1 cytotoxic T (Tc1) cells to attract them into the lungs. Tc1 cells, through the release of perforin and granzyme B, induce apoptosis of type 1 pneumocytes, thereby contributing to emphysema. IFN released by Th1 and Tc1 cells then stimulates further release of CXCR3 ligands, resulting in a persistent inflammatory activation.
B-cells
B-cells have an important role in allergic diseases, including asthma, through the release of allergen-specific IgE which binds to high-affinity Fc receptors (FcϵRI) on mast cells and basophils and to low-affinity Fc receptors (FcϵRII) on other inflammatory cells, including B-cells, macrophages and possibly eosinophils [66]. The Th-2-type cytokines IL-4 and IL-13 induce B-cells to undergo class switching to produce IgE. Blocking IgE with a monoclonal antibody, omalizumab, reduces the response to allergens, airway inflammation and asthma exacerbations, indicating that IgE drives allergic inflammation in asthma [67]. In both atopic asthma and non-atopic asthma, IgE may be produced locally by B-cells in the airways [68].
Interestingly, IgE secretion is not observed in patients with COPD; however, in the peripheral airways of patients with more severe disease, there is a marked increase in the number of B-cells, which are organised into lymphoid follicles that are surrounded by T-cells [13]. The class of immunoglobulin they secrete and how they are regulated is currently unknown, but they may be activated by bacterial or viral antigens as a consequence of the chronic bacterial colonisation or latent viral infection in the airways of these patients. Alternatively, it has been suggested that COPD might have an autoimmunecomponent with the development of new antigenic epitopes as a result of the tissue damage induced by cigarette smoking, oxidative stress or chronic bacterial infection [16, 69]. CD4+ T-cells isolated from the lungs of patients with severe emphysema are oligoclonal, which is consistent with antigenic stimulation by infective organisms or autoimmunity [70]. In a mouse model of emphysema induced by tobacco smoke, an autoimmune mechanism has been proposed with a role for neutrophil-elastase-specific antibodies [71].
Dendritic cells
Dendritic cells (DCs) have an important role in asthma as regulators of Th-2 cells and in the presentation of processed peptides from inhaled allergens to Th-2 cells [72]. They are not only involved in the initial sensitisation to allergens, but also in driving the chronic inflammatory response in the lungs and therefore provide a link between allergen exposure and allergic inflammation in asthma. The cytokine thymic stromal lymphopoietin (TSLP), which is secreted in large amounts by epithelial cells and mast cells of asthmatic patients [73, 74], may have a critical role in the maturation of myeloid DCs and the recruitment of Th-2 cells to the airways by inducing the release of CCL17 (also known as TARC) and CCL22 (also known as MDC), which bind to CCR4 that is selectively expressed by Th-2 cells [75].
Cigarette smoking is associated with an expansion of the DC population and with a marked increase in the number of mature DCs in the airways and alveolar walls of people who smoke [76]. The role of DCs in COPD is currently unclear as there are no obvious antigenic stimulants as there are in asthma; however, α-glycoprotein, isolated from tobacco, is known to have a powerful immunestimulatory effect. Alternatively, a recent electron microscopy study has demonstrated a decrease in DCs is the airways of patients with COPD who smoke compared with smokers without airway obstruction, suggesting that they do not play a key role in COPD [77]. Conversely, another study shows a clear increase in the numbers of dendritic cells in COPD patients, indicating that they may play a role in linking innate and acquired immunity [78].
Similarities between asthma and COPD
Although the inflammatory and immune mechanisms of asthma and COPD described above are markedly different, there are several situations where they become more similar and the distinction between asthma and COPD becomes blurred (table 2).
Severe asthma
Although only about 5% of the asthmatic population develop severe disease, such cases account for over half of the healthcare spending in asthma and they are poorly controlled by currently available therapies [79]. The inflammatory pattern occurring in severe asthma becomes more similar to that occurring in COPD than in mild asthma, with increased numbers of neutrophils in the sputum, together with increased amounts of CXCL8 and tumour-necrosis factor [80], increased oxidative stress and a poor response to corticosteroids as is observed in patients with COPD (table 2). Moreover, whereas in mild asthma Th-2 cells predominate, in more severe disease there is a mixture of Th-1 and Th-2 cells present in bronchial biopsies, as well as more CD8+ T-cells and this more closely resembles the immune-cell infiltration seen in COPD [46, 64, 65]. The neutrophilic inflammation seen in cases of severe asthma may by induced by IL-17 production by Th-17 cells, which induces the release of the neutrophilic chemokine CXCL8 from airway epithelial cells [54, 55]. A neutrophilic pattern of inflammation, with high levels of CXCL8, is also found in the sputum of asthmatic individuals who smoke [81]. Similar to patients with severe disease or COPD, these individuals also have a poor response to corticosteroids, even if given orally in high doses.
Reversible COPD
Approximately 10% of patients with COPD patients have a reversibility of bronchoconstriction, showing >12% improvement in lung function as assessed by forced expiratory volume in 1 s (FEV1), and therefore behave more like asthmatics. Furthermore, compared with most patients with COPD, these patients more frequently have eosinophils in their sputum, an increase in exhaled nitric oxide and respond better to corticosteroid treatment, all of which are features of asthma [82, 83]. It therefore seems likely that these patients have concomitant asthma and COPD.
Acute exacerbations
Acute exacerbations (worsening of symptoms) occur in patients with asthma and COPD, and are a major cause of patient suffering and medical expenditure [84, 85]. Exacerbations in asthmatic individuals are usually triggered by upper respiratory tract infections, such as with rhinoviruses, and less commonly by inhaled allergens and air pollutants, whereas exacerbations in patients with COPD are usually triggered by either bacteria or viral infections. In both diseases, exacerbations are associated with a further increase in airway inflammation, increased numbers of cells infiltrating the lungs and higher concentrations of inflammatory mediators than are present in the stable state. However, there may also be changes in the pattern of inflammation. In exacerbations of asthma triggered by viruses, there can be increases in the numbers of neutrophils, as well as of eosinophils [84], whereas in COPD exacerbations, particularly those due to viruses, there may be an increase in eosinophil numbers [86]. So, during episodes of disease exacerbation, the pattern of inflammation becomes similar in COPD and asthma.
Footnotes
This article is based on a post-graduate course from the Barcalona 2010 ERS Annual Congress
Competing interests
P.J. Barnes has received funding for lectures and research from Astra Zeneca, GlaxoSmithKline, Boehringer Ingelheim, Novartis and Pfizer.
- ©ERS 2011
References










