





Abstract
Educational aims
-
To understand which techniques are available to monitor early lung disease in cystic fibrosis, recognise difficulties inherent to each measure, and describe the use of tests in clinical practice or interventional trials.
Summary It is a particular challenge to detect and monitor lung disease in children younger than 6 years old. Various methods exist that can reveal abnormality in young children, but each technique presents difficulties on performing the test or interpreting results. Most children with cystic fibrosis are now diagnosed shortly after birth, but it is still unclear how to evaluate new therapies, or at what age to initiate them. This review summarises current options for monitoring early disease, limitations of individual techniques and how evidence to date influences their use in research and clinical practice.
Key points
-
Lung disease remains the major cause of morbidity and mortality in cystic fibrosis
-
Despite clear improvements in outcomes such as nutrition, the benefit of early diagnosis by newborn screening on pulmonary health in cystic fibrosis has yet to be proven.
-
Early lung disease is challenging to detect and monitor in young children with cystic fibrosis, but there is an urgent requirement to define useful outcome measures in this population, particularly as new treatments emerge.
-
Many techniques are available, but difficulties with measurement or interpretation must be recognised when appraising past studies, considering their use in practice or as endpoints in clinical trials.
-
There is no superior monitoring test at present, but emerging evidence from longitudinal studies in children with cystic fibrosis will help define which are most useful.
Introduction
Significant advances have been made in cystic fibrosis (CF) care in the last decade. Earlier diagnosis, care in specialist centres and more intensive therapy have contributed to an increased median age of survival from 23 years in 2002 [1] to 43.5 years in 2012 [2] in the UK. Newborn screening has enabled intervention shortly after birth, and considerable improvement has been seen in outcomes such as nutritional status [3]. The impact on pulmonary health however is less striking [4]. Progressive lung damage may be delayed but inevitably still occurs and remains the major cause of morbidity and mortality in CF.
Children with CF often appear well during the early years, but there is compelling evidence that pulmonary inflammation, infection and structural changes take hold alarmingly soon after diagnosis [5–7]. What is most concerning is that there are often no easily detectable markers of disease and by the time symptoms develop, irreversible damage may have occurred. Early infection and inflammation also seem to impact on future health, with evidence that lung function does not return to baseline after an exacerbation, even in infancy [8].
Earlier studies of monitoring measures are based on children diagnosed clinically with CF. Prompt diagnosis in newborn screened infants now offers a unique opportunity to determine those who get earlier disease, identifying candidates for more aggressive treatment or interventional trials. This becomes increasingly important as treatments targeting the basic defect in the cystic fibrosis transmembrane conductance regulator (CFTR) protein are developed. Which markers are most informative remains an area of contention and investigation in infants and young children involves practical, ethical and regulatory difficulties [9]. This review focuses on the most promising methods to detect lung disease in children with CF, and what must be considered when evaluating their use (table 1).
Infant lung function techniques
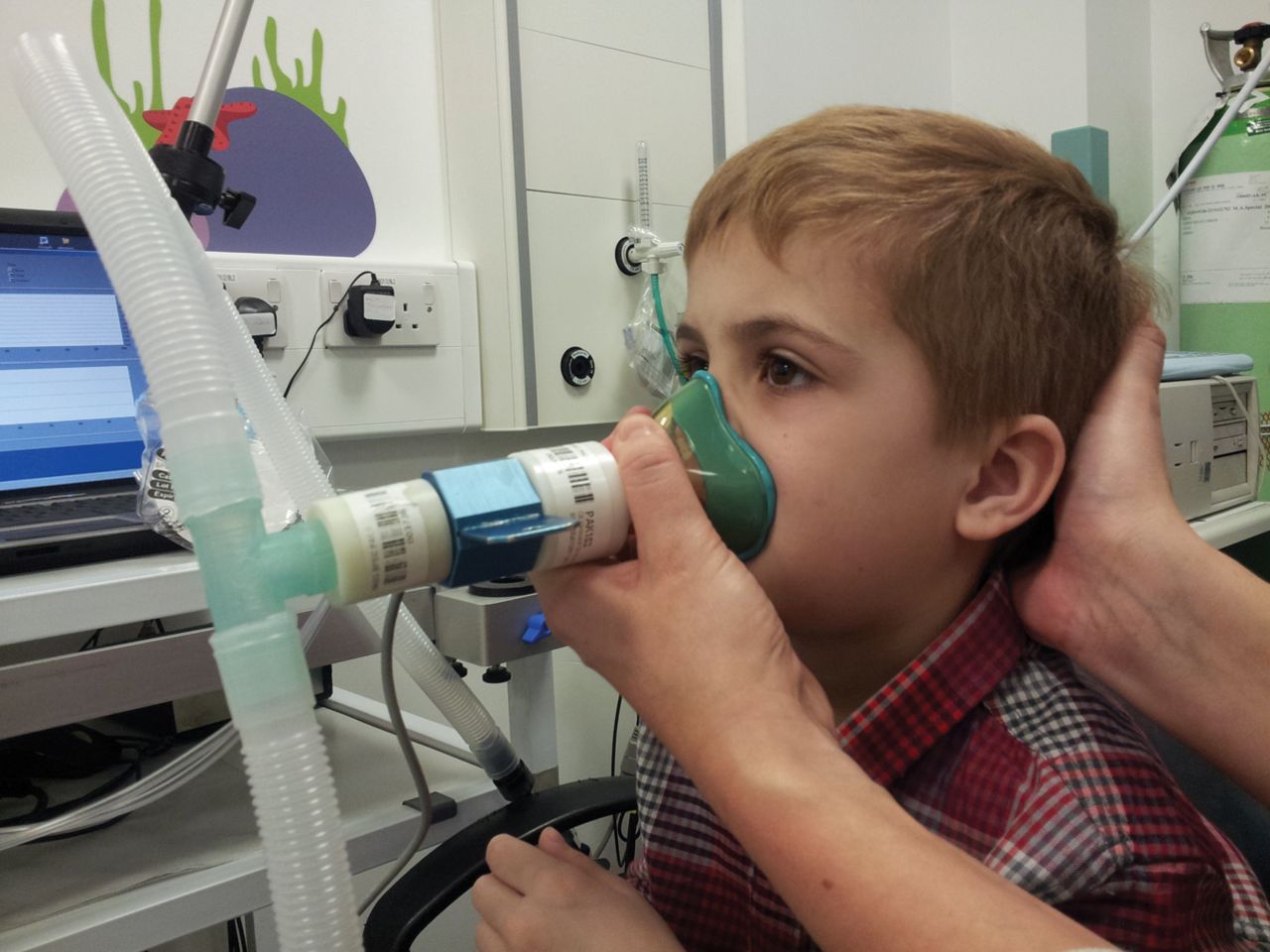
Infant lung function testing is a noninvasive option when monitoring CF lung disease throughout infancy. A number of methods and measures are available. The two that give the most useful information in early CF disease are the raised volume rapid thoraco-abdominal compression (RVRTC) technique (fig. 1) and full body plethysmography, both normally performed under sedation. In the RVRTC technique [10], the lungs are inflated towards total lung capacity with three to five breaths until an inspiratory pause is produced. A vest wrapped around the infant's chest is quickly inflated and forces expiration to residual volume [11], giving measures of forced expiratory flow and volume. The forced expired volume in 0.5 seconds (FEV0.5) is normally reported as the lungs may empty in infants before 1 second has passed.

The raised volume technique in a sedated infant. Figure reproduced with permission from J. Stocks.
Lung volumes are measured in a plethysmograph when an infant breathes against a closed shutter [12]. Changes in pressure and volume inside the closed system are used to calculate unknown volumes of the lungs. Functional residual capacity (FRC), the volume of gas left in the lungs after a tidal expiration, is the most commonly reported measure.
Commercial devices are available for the measurement of infant lung function, and standards for testing are available for both the RVRTC technique [13] and plethysmography [12]. Important considerations in any measurement of lung function are feasibility, reproducibility and choice of reference data to interpret results. In a recent 10-centre study of infants with CF, acceptability rates were relatively high overall, at 72% for RVRTC measures and 89% for plethysmographic FRC [14]. However, despite meticulous training and quality control, feasibility varied significantly between sites depending on experience. The reproducibility of these tests is summarised in table 2.
Data on longer-term reproducibility are scarce as repeated test occasions, and therefore sedation, of the same infant would be required. The need for sedation also means that reference data from healthy infants are limited. The Jones reference equations are frequently used for RVRTC values, but are derived from only 155 infants measured across two centres using noncommercial equipment [16]. These equations have been shown to grossly overestimate abnormality when applied to data measured on a different commercially made device [17]. Equations for FRCpleth have recently been published from 153 infants [18], but again should be considered equipment specific. Use of contemporaneous healthy controls or equipment specific equations in studies is essential for meaningful interpretation of data [19] and important to appraise in research reports.
Studies of infant lung function in children with CF
Two main groups have described the evolution of lung function, alongside other measures, in infants with CF: the AREST-CF (Australian Respiratory Early Surveillance in CF) group and the LCFC (London Cystic Fibrosis Collaboration). They have both found that measures from RVRTC and plethysmography distinguish patients with CF as a group from healthy infants [5, 14, 20–23], but there is considerable overlap in individuals. The age at which lung function becomes abnormal and its evolution in infancy varies between groups. In the AREST-CF cohort, lung function was initially reported as normal in the first six months of life [20], but was found to be abnormal before this age in a subsequent study [24]. A significant decline of lung function in infancy was found despite early diagnosis and treatment, with FEV0.5 2 z-scores below the mean by one year. By 2 years this deficit was even more abnormal at -4 z-scores, despite most infants being asymptomatic of disease. In the newborn screened infants of the LCFC cohort, lung function was abnormal by 3 months of age [5]. In contrast to the AREST-CF findings, lung function either improved or remained stable at 1- and 2-year followup visits [25, 26]. Importantly this differed from earlier reports in clinically diagnosed infants with CF, for whom abnormal lung function progressed after diagnosis [22], suggesting that earlier instigation of treatment in newborn screened infants may help to preserve lung function in infancy. It is unclear why reports differ so greatly between these two similar cohorts of children. Different treatment protocols could have an effect. It may also be explained in part by the use of reference data to interpret results. The AREST-CF group used historical control data derived from children tested on different equipment to calculate z-scores; whereas the London group studied healthy control subjects tested under the same conditions and derived equipment specific reference data [27].
Regardless of mode of diagnosis, abnormal infant lung function tests correlate with structural lung changes on computed tomography (CT) [28] and inflammation detected by bronchoalveolar lavage (BAL) [24, 29]. A greater decline in lung function is seen in those infected with respiratory pathogens [24, 30, 31], providing evidence for aggressive treatment of infection in infancy.
An important question is whether infant lung function measures can predict those in older children, and identify an “at risk” group that can be targeted for intervention. In clinically diagnosed infants with CF, abnormal results predicted deficits in lung function later in infancy, at preschool age and at school age [22, 25, 30, 32, 33]. Data in newborn screened infants are scarce, with one study reporting lung function in infancy and pre-school spirometry [31]. In this study, no clear longitudinal association was seen, and there were significant inter-individual differences in tracking of lung function.
Infant lung function measures have been used in interventional studies, but numbers of participants are often small. In 11 infants, lung function was shown to improve after antibiotic treatment of a respiratory exacerbation [8], showing potential to detect change with treatment. RVRTC measures have been used in the Infant Study of Inhaled Hypertonic Saline in Cystic Fibrosis (ISIS) trial, with FEV0.5 showing a small improvement with treatment [34]. Large numbers of infants are likely to be needed if infant lung function is to be used as a meaningful outcome measure. Data from a recent multi-centre observational study estimated that 150 infants would be needed in each treatment arm to sufficiently power an interventional trial to detect a significant treatment effect [35]. Other considerations when using infant lung function techniques are the need for sedation, time-consuming procedures, intense training of staff and expensive equipment [27]. It currently remains a measure confined to specialist centres, and use in clinical practice or as an outcome measure is limited for these reasons.
Pre-school spirometry
With regards to lung function, the time between ages 2 and 5 years are often referred to as “silent”; in which infant lung function testing is no longer feasible and children cannot reliably perform conventional spirometry. However, in an age-appropriate, relaxed and enthusiastic environment, a reproducible forced expiratory manoeuvre can be obtained in a preschool child. Training with games using bubbles or whistles and computerised incentives during testing can help achieve maximal peak flow and vital capacity (fig. 2) [36].

Examples of computerised incentives used in spirometry. a) Blowing out candles incentive to encourage maximal peak expiratory flow. b) Bowling game to encourage expiration to forced vital capacity. Image reproduced with permission from CareFusion Corporation.
Many commercial devices are available to measure pre-school spirometry. Guidelines for testing and modified quality control criteria are available [37, 38] and are summarised in table 3.
Quality control should include real-time inspection of flow–volume and volume–time traces. A full expiration may be reached before one second in some children, so FEV0.5 and FEV0.75 should be recorded in addition to FEV1 [38]. The largest FEVt and FVC should be reported even if not taken from the same manoeuvre, and forced expiratory flows are taken from the manoeuvre with the largest sum of FEV0.5 and FVC [37]. In an experienced laboratory, acceptable spirometry can be obtained in over 75% of pre-school children, and success rates increase with age [39, 40]. Between-test reproducibility is good for FEV0.5, FEV1 and FVC (intraclass correlation coefficient (ICC) 0.85–0.92) and moderate for FEF25–75 (ICC 0.60) [39].
Current software programs for commercial devices use a combination of reference equations to cover age ranges. This may lead to over- or under-diagnosis of abnormality on certain test occasions [41] with obvious clinical consequences. The recently published Global Lung Function Intitiative “all age” equations are based on measurements of over 4000 children, span from 3 to 95 years and consider ethnic group [42]. They cover transition from child to adulthood more smoothly to aid accurate interpretation of results. The range (or standard deviation) of normality varies greatly with age, especially in younger children, and standard z-scores should be reported rather than percent predicted values [35] to avoid misinterpretation.
Previous studies of spirometry in children with CF
Spirometric values are on average lower in preschool children with CF when compared with controls, but only a small proportion (9–36%) have an FEVt below the normal range [32, 36, 40, 43–45] as disease is often mild. FEVt has also been reported as normal when other markers indicate disease [40, 46] and is therefore relatively insensitive to abnormality in this age group. Forced expiratory flows are more reduced than volumes [32, 44] and may be a more useful measure. When compared with other measures of lung function at pre-school age, a multicentre study showed that spirometry was more sensitive in detecting abnormality in CF than inductance plethysmography or forced oscillation [39].
With regards to predicting lung function in later life, most data are available in clinically diagnosed children. Two studies have reported longitudinal data from infancy [24, 30]. Abnormalities appear to track from pre-school to school age, and worsen with age [33]. Data in new-born screened children are awaited.
The familiarity of clinicians with parameters reported in spirometry makes it an attractive measure for use in practice. If performed accurately in an experienced centre, spirometry is repeatable and may be used to monitor progress over time or with intervention. However its use is limited by insensitivity to very early changes in CF lung disease, as large numbers would be required to detect any significant treatment effect.
Inert gas washout
Inert gas washout was first described in the 1940s and examines gas mixing in the lung [47]. In young children, multiple- (rather than single-) breath washout (MBW) is commonly used as it can be performed during tidal breathing when an infant is sedated, or pre-schooler distracted by watching television. The technique involves recording the decline in concentration (washout) of an inert tracer gas from the lungs. Tracer gases must have low solubility in the blood and not participate in gas exchange, and those commonly used are sulphur hexafluoride (SF6), helium and resident nitrogen. With SF6 or helium, the inert gas is washed into the lungs, i.e., inspired until an equilibrium concentration is reached (fig. 3). The gas supply is then disconnected at end-expiration and the washout recorded until an end concentration of the inert gas is reached, commonly 1/40th of the starting concentration. Nitrogen washout is performed in a similar way, but no wash-in period is required as resident nitrogen can be washed out by inspiring 100% oxygen. A mouthpiece is used in older subjects, but in young children a mask sealed with putty is applied to the face to ensure no leak of gas during the washout and minimise dead space of the mask.

A pre-school child during the wash-in phase of multiple breath washout. Image copyright P. Aurora.
Functional residual capacity can be calculated from the washout (fig. 4). The initial concentration of tracer gas is known at the beginning (Cinit) and end (Cend) of the washout, as is the total volume of gas expired. FRC can therefore be calculated using: FRC = volume of gas expired/((Cinit) - (Cend).

Example of a washout recording. The black trace is flow and green trace SF6 concentration.
FRC measured by MBW includes only the volume that equilibrates with tracer gas during tidal breathing (i.e. not trapped gas) and may be a smaller volume than that calculated by plethysmography, which measures total thoracic gas [48]. In diseased airways, inflammation, mucous plugging and airway wall damage will alter gas mixing and it becomes inhomogeneous [49]. The lung clearance index (LCI) is a simple measure commonly reported from washout recordings to describe overall ventilation homogeneity. It reflects the number of times the FRC must be “turned over” to bring the tracer gas to reach the set end-concentration [11], and is an index of this turnover volume divided by the measured FRC. In disease, the number of FRC turnovers to clear the tracer gas, and therefore LCI, are increased: LCI = cumulative expired volume/FRC.
Other measures that can be reported from the washout include progression analysis of the alveolar plateau (phase III slope) of expired breaths. This gives more information about the relative contributions of conducting or acinar airways to ventilation inhomogeneity, but the feasibility of these measures is limited in preschool children, and particularly infants, due to difficulty in analysis.
Guidelines for technique and quality control in MBW are available [37, 50]. A fast gas analyser is required and the gold standard at present for measurement is mass spectrometry. Use therefore is limited to specialist centres, but validation of commercial devices in children is currently underway. It is highly feasible, with success rates of over 80% in sedated infants and preschool children [21, 40]. There is greater within- and between-test variability of LCI in infants [51], but within-test coefficient of variation in pre-schoolers is only 5.2% [40]. LCI is independent of age in older children and adults, making it a useful longitudinal measure; however, the upper limit of normal is higher in infants and pre-school children. Reference equations in children from 497 subjects have been published using SF6 measured by mass spectrometry [52] but are tracer gas and device specific. Comparisons to healthy controls are again essential in studies using other testing methods.
Previous studies of MBW in children with CF
In the LCFC cohort, Lum et al [21] reported raised volume tests and LCI in infants diagnosed with CF by newborn screening and controls up to 1 year of age. LCI was abnormal in 56% of infants with CF. There was correlation with RVRTC and FRCpleth measures in some but not all of these infants [21]. RVRTC measures and LCI seemed to be complementary in detecting early disease; both were abnormal in 41%, and each measure identified a further 15% with abnormal results. By 1 year LCI remained stable, but abnormal LCI at 3 months did not predict those with abnormal LCI at 1 year [25]. Longitudinal data to later childhood in newborn screened cohorts are awaited. LCI is abnormal in infants infected with Pseudomonas aeruginosa and correlates with inflammatory markers from BAL fluid [53]. Only weak associations have been found between LCI and structural changes on CT in infancy [54].
Reports in pre-school children contrast to those from infancy, but most reports are in those clinically diagnosed with CF. At this age, LCI clearly distinguishes those with CF from healthy controls [40] and in this report, 73% of children with CF had abnormal LCI, 47% abnormal plethysmography and only 13% abnormal spirometry. LCI appears as sensitive as computed tomography when detecting abnormality at school age [55]. Correlation between these two measures in preschool children is yet to be reported. Importantly, increased LCI at preschool age has been shown to predict abnormal spirometry in school-age children with CF [43] and in a group of patients studied from 6 to 20 years, LCI was an earlier predictor for the progression of lung disease than FEV1 [56].
LCI has detected a treatment effect in trials of dornase alpha [57], inhaled hypertonic saline [58] and ivacaftor [59] in children with otherwise normal lung function. Disease in infants may be too subtle for MBW to detect or predict abnormality but, in preschool children, it has great potential as an outcome measure in trials and can inform clinical practice. It is still necessary to show that regular monitoring with LCI results in improved outcome [60] and it is currently limited to centres with facilities to perform the test. Its use can become more widespread with validation of commercial equipment.
Other measures of preschool lung function
Plethysmography is commonly used to measure lung volumes and airway resistance in adults, but involves breathing against a closed shutter which younger children find difficult to tolerate. An alternative plethysmographic measure is specific airway resistance (sRaw)(fig. 5), which is the product of airway resistance and FRC, and can be measured during tidal breathing [61]. It has been measured successfully in children from 2 years of age [62]. Preschool subjects breathe through a mouthpiece attached to a pneumotachograph, wearing a nose-clip with their hands on their cheeks. This can be demonstrated to the child in steps before recording to improve success rates, which are reported to be between 77 and 81% [40, 45]. Airflow and plethysmographic pressure change are recorded and specific resistance can be calculated.

A pre-school child performing tidal breathing manoeuvres for the measurement of sRaw inside a plethysmograph. Image copyright P. Aurora.
As sRaw increases with higher FRC or airway resistance, it is a useful measure in children with CF, as both can be raised in obstructive lung disease or hyperinflation [15]. Reference equations have been recently published using measurements from nearly 3000 children as part of the Asthma UK initiative [63]. Significant methodological differences between centres when measuring sRaw were noted in this study, and consensus for appropriate breathing frequency and other quality control factors are yet to be decided, with guidelines for standard operating procedures urgently needed. Two main studies have reported sRaw measures in children with CF [40, 45]. In both studies sRaw was significantly higher than healthy controls. sRaw was persistently abnormal over a 4-year period and identified those with abnormal lung function earlier than spirometry [45]. A positive correlation has been seen with lung clearance index [40]. sRaw has potential to monitor early CF lung disease, but standard operating procedures are crucial if this is to be developed.
Other available measures of pre-school lung function are the forced oscillation (FOT) and the interrupter technique. They are also both tidal breathing measures. Forced oscillation measures respiratory mechanics, and involves the application of an external sound wave to the respiratory system. The resulting pressure and air flow response is then analysed and respiratory resistance (Rrs), reactance (Xrs), and impedance calculated. There are conflicting reports of FOT in young children with CF, with some studies showing abnormal Rrs and Xrs [64, 65] and others normal values [39, 45]. No relationship was found in these studies between FOT measures and respiratory infection.
The interrupter technique involves measuring pressure at the mouth over a series of brief airway occlusions during tidal breathing. It assumes that alveolar pressure equilibrates with mouth pressure during the occlusions. Interrupter resistance (Rint) is the ratio of pressure at the mouth measured after an occlusion and flow just before it [66]. Most studies in children with CF have shown that Rint does distinguish those from healthy controls and remains stable over time, despite other parameters indicating worsening disease [45, 67]. Further reports will help define the role of these alternative tests in monitoring early CF lung disease.
Imaging
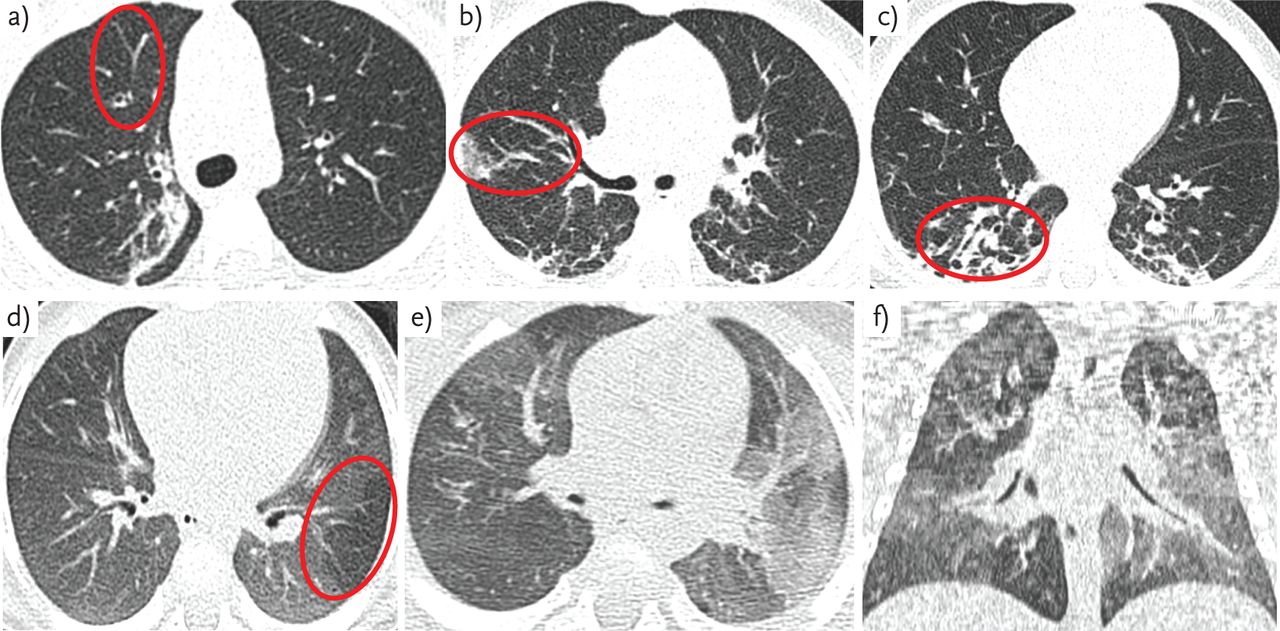
Progressive structural changes, particularly bronchiectasis, are the hallmark of lung disease in adults with CF [68]. These changes can also be apparent in children, and histological specimens have shown bronchiectasis and mucus plugging as early as 4 months of age [69]. CT is more sensitive than chest radiography in detecting CF lung disease in children [70] and advances in imaging techniques provide the opportunity to image structural changes in ever increasing detail (fig. 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Examples of CT images in CF showing a–c) bronchial wall thickening/dilatation and atelectasis; and d–f) air trapping.
As young children are unable to breath-hold sufficiently, scans are normally performed under general anaesthesia. Scoring of abnormality differs with lung volume, so standardisation of lung inflation during imaging is particularly important. Images taken in inspiration and expiration in newborn screened children (aged 1–5 years) have been compared, with bronchiectasis reported in 30% of inspiratory scans, but only 9% of expiratory scans, and 6% in tidal breathing [71]. Recent studies in newborn screened infants use strict ventilation protocols before imaging, and administer a “breath-hold” (usually at 25 cm H2O) during the inspiratory scan [7, 72]. Concerns over radiation exposure in such young children have led to the development of low-dose scanning techniques [73], and “limited slice” protocols taking 3–6 images at standard anatomical positions in inspiration and repeated in expiration have been used [74]. However, due to the irregular distribution of lung disease in CF, limited slice protocols may neither capture all areas of structural abnormality [75] nor provide adequate resolution to quantify early disease [28].
Various scoring systems are available to quantify structural abnormalities. Modified CT scores have been used in recent studies of new-born screened infants with CF, in the AREST-CF and LCFC cohorts [7, 72] but the validity of scores in infants and young children needs clarification.
Previous studies of CT in children with CF
Structural changes that can be observed on CT in children less than 5 years of age with CF include bronchial wall thickening, air trapping and bronchiectasis, but changes are often mild [70]. Abnormalities have been detected by CT in newborn screened infants shortly after diagnosis in the AREST-CF cohort [7]. At 3 months of age 18.6% had bronchial dilatation, 45% bronchial wall thickening and 66.7% gas trapping, giving a total of 80.7% of infants with an abnormally scored CT. Most children had no clinical symptoms of disease. The proportion of children with bronchiectasis increased with age from 8.5% in the first year of life to 56% in 4–6 year olds [76]. Once present, bronchiectasis was seen to persist in nearly 75% of children and became more severe in over half [77], despite early diagnosis and treatment. In 27%, some improvement was seen, suggesting that some changes reversed despite being described as bronchiectatic. Progression of bronchiectasis was associated with bacterial infection, particularly with P. aeruginosa [76], and inflammation detected on BAL fluid [77]. The marker most associated with the presence, extent and progression of bronchiectasis was neutrophil elastase [7, 77]. It is important to note that scans in these reports were scored by one individual, and the reproducibility of this scoring method is therefore unknown.
In contrast to these findings, the LCFC cohort studies found CT to be abnormal by 1 year of age but changes were very mild [72]. Using a similar scoring system, most scans were scored as normal [78]. Only poor-to-moderate inter- and intra- observer agreement was reported despite two experienced senior paediatric radiologists undergoing thorough training in the score. This brings into question the accuracy of using current scoring systems in this young population (with such subtle disease) that to date have only been validated in older children. Automated quantitative scoring systems have potential to be a more reproducible scoring measure, but also require validation.
Abnormalities in early childhood appear to predict lung disease progression, with abnormal CT score associated with significantly worse lung function in later life [79]. CT scores have been shown to improve after treatment for an exacerbation [80], dornase alpha [81] and inhaled tobramycin [82] and its use as an outcome measure in young children is promising. However it is difficult to justify regular surveillance CT in such a young population who already have a high lifetime radiation exposure and are surviving longer, particularly until robust scoring systems for CT imaging are developed. Magnetic resonance imaging (MRI) has been shown to detect changes in adults with CF [83, 84] but is limited at present when studying subtle early small airway changes in younger children. Hyperpolarised helium MRI is more sensitive for the detection of lung abnormalities in CF [85], but use is limited by cost and availability. As functional and structural techniques develop the use of MRI in this population may increase.
BAL
As young children may be unable to expectorate sputum, BAL is a useful alternative to detect infection and quantify inflammatory biomarkers in CF, particularly when performed alongside other measures of early disease. An ERS taskforce statement was published in 2000 giving guidelines for obtaining, storing and processing lavage fluid [86]. Centres researching the role of BAL in young children with CF have developed protocols to standardise test procedures, but there is still potential for technique to vary. It is recommended that lavage is performed in the most affected lobe if detected by prior imaging, or the right middle lobe in the presence of diffuse disease. However, as CF lung disease is “patchy”, lavaging one lobe does have the potential to miss areas of disease [87]. There is no consensus on the number or volume of aliquots that should be taken at present. There may be differences in volumes and dilution of fluid retrieved from each patient, and values of biomarkers vary in sequential aliquots. During analysis, individual or pooled aliquots can be studied, but are likely to yield different results [87]. There are many potential assays available which vary between laboratories, and data on reproducibility of markers are scarce. The European Cystic Fibrosis Society Clinical Trial Network Standardisation Committee have published a review on BAL markers and report neutrophil count or percentage, neutrophil elastase, interlukin-6 and interlukin-8 as the most useful currently in CF [87]. Limitations of this procedure in young children also include its invasive nature and need for general anaesthesia. Minor adverse events such as fever are common but otherwise the procedure is reported as relatively safe [88].
Previous studies of BAL in children with CF
Inflammation and infection have been identified at diagnosis in newborn screened infants [7, 89] and persists to the preschool years [76]. There is emerging evidence that inflammation may occur before infection in CF [7] and inflammation (absolute cell and neutrophil count) has been detected in infants at 4 months with negative microbiology [89]. Many studies report increased inflammation with infection [7, 90–92]. In a recent longitudinal study of newborn screened infants with CF [7], raised free neutrophil elastase activity at 3 months of age was the strongest predictor for bronchiectasis diagnosed by CT, with the odds of persistent bronchiectasis increased seven-fold at 12 months and 4-fold at 3 years [93]. There is a correlation between increased inflammation and abnormal infant lung function tests [24].
Markers in BAL have been studied in trials of tobramycin inhaled solution [94] and recombinant human DNase [95], but no clear change in any one marker was found. BAL is also a useful clinical tool when infection is suspected with otherwise negative microbiology. However, in a trial of BAL-directed therapy compared with standard treatment in children infected with P. aeruginosa (from diagnosis to 5 years of age), no difference in the rate of pseudomonas eradication or CT score between the two approaches was found [96]. BAL remains useful in the early stages of trials to describe pathophysiological processes in CF [87] and free neutrophil elastase activity detected in infants appears an important predictor for more severe lung disease.
Conclusion
The evidence for early inflammation and infection in young children with cystic fibrosis continues to grow, as does our understanding of the early disease processes occurring in the lungs of children with CF. In infancy, it appears that lung function testing and LCI may have a complementary role in identifying abnormality, and further reports will help define whether they can be used to predict future disease in children diagnosed by newborn screening. The role of imaging as an outcome measure in young children will become clearer as techniques advance and more robust scoring systems are developed. In preschool children, MBW is a feasible test sensitive to pulmonary disease in CF, and its role in interventional studies and clinical care will increase as commercial devices are validated in this age group. The opportunity to identify and treat disease in children before permanent lung damage occurs remains an exciting possibility, and further research in this area will bring us closer to this goal.
Educational questions
Which of the following statements are true?
-
Regarding infant lung function
-
Guidelines for standards of testing are available for both the RVRTC technique and plethysmography
-
Reference data are derived from testing healthy control subjects
-
Published reference equations can be applied to any test device
-
After appropriate training in technique and quality control, feasibility varies little between centres
-
In infants with CF a greater rate of decline is seen in those infected with respiratory pathogens
-
-
Lung function measures obtained during tidal breathing include
-
Specific airway resistance
-
Respiratory impedance
-
Multiple breath washout
-
Forced vital capacity
-
Interrupter resistance
-
-
Regarding imaging in children with CF
-
A robustly validated CT scoring system is available for use in infants
-
Scoring of abnormality varies little when images are taken at different lung volumes
-
Bronchiectasis on CT imaging has been reported at diagnosis in infants
-
Once detected, bronchiectasis is persistent in most children
-
Hyperpolarised helium MRI is less sensitive than conventional MRI techniques in detecting abnormality
-
-
Bronchoalveolar lavage in children with CF
-
Should be performed in the right middle lobe when disease is diffuse
-
Has been shown to improve rates of Pseudomonas aeruginosa eradication when treatment is directed by lavage results
-
Can predict a decreased risk of the development of bronchiectasis if free neutrophil elastase is detected at 3 months of age
-
Yields similar values of inflammatory markers in individual and pooled aliquots of lavage fluid
-
Results in frequent major adverse events
-
-
With respect to inert gas washout
-
Single breath washout is a useful measure in preschool children
-
Published reference equations are available for the interpretation of LCI
-
Commercial devices are validated in young children
-
Abnormal LCI detected at diagnosis of CF persists throughout infancy in most children
-
MBW is more sensitive than spirometry in the detection of abnormality in pre-school children with CF
-
Answers to educational questions
-
a, b, e
-
a, b, c, e
-
c, d
-
a
-
b, e
Footnotes
Statement of Interest
None declared.
- ©ERS 2014
Breathe articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
REFERENCES










