





Abstract
Educational aims
-
To describe the emerging genetics of primary ciliary dyskinesia (PCD) and the heterogeneity of the disease
-
To highlight the clinical symptoms and signs suggestive of PCD that should lead to consideration of diagnostic testing
-
To highlight the difficulties in diagnosing PCD emphasising the need for specialist diagnostic centres
-
To discuss current treatment strategies and highlight the lack of an evidence base for these
Summary Primary ciliary dyskinesia (PCD) is a rare heterogeneous genetic disorder affecting ciliary function. Genes coding for various ciliary structural proteins or cytoplasmic proteins responsible for the assembly of cilia can be mutated resulting in abnormal ciliary function. However, despite the diversity of genotypes that can cause PCD the clinical phenotypes of PCD are all remarkably similar. The main clinical symptoms are caused by a lack of mucociliary clearance. Worryingly many patients are diagnosed late despite their classical, lifelong symptoms of a daily wet sounding cough and rhinosinusitis. Even when PCD is suspected, poor access to specialist diagnostic centres may delay diagnosis. Currently, diagnostic testing includes screening of nasal nitric oxide, followed by nasal brushing to obtain ciliated epithelial strips for high-speed video analysis of ciliary function. This is typically followed by transmission electron microscopy and in difficult cases by ciliated cell culture. Emerging tests including immunofluorescence and genetic examination are the focus of intense research and are likely to feature more in the future. Despite a greater understanding of the pathogenesis of PCD and improved diagnostic testing, management strategies are currently based on expert opinion with little, if any, evidence base.
Key points
-
Mutations causing PCD have been found in almost 30 genes and it is likely many more will be discovered. Despite this genetic heterogeneity, the clinical features are remarkably similar.
-
The age at diagnosis is high across Europe; early diagnostic testing should be prompted by symptoms that include unexplained respiratory distress in term neonates, a lifelong wet sounding cough, chronic rhinosinusitis and hearing problems due to glue ear.
-
Diagnosis of PCD is complex and should be carried out in specialist centres. Ideally, specialist respiratory centres should coordinate management of patients.
Introduction
Primary ciliary dyskinesia (PCD) is a rare autosomal recessive genetic condition. Although the estimated prevalence is between 1 in 15 000 and 1 in 30 000 [1–3] a recent study found a prevalence of 1 in 2200 in a well-defined, highly consanguineous British Asian population [4]. Studies suggest that PCD is still underdiagnosed and that factors contributing to this may include lack of recognition of the clinical presentation, lack of diagnostic centres and cases that have a normal ultrastructure on transmission electron microscopy [2, 5].
PCD patients were initially described by Kartagener and Stucki [6] who noted the familial occurrence of chronic sinusitis, situs inversus and bronchiectasis. In 1976, Afzelius [7] noted the connection between chronic respiratory problems, male sterility, situs inversus, immotile cilia and abnormal ciliary ultrastructure. It was subsequently discovered that the same clinical phenotype was also seen in patients whose cilia were motile but dyskinetic and the name of the condition changed from “immotile cilia syndrome” to “primary ciliary dyskinesia” [8].
Cilia
Cilia are highly evolutionary conserved complex organelles [9]. Motile cilia drive flow along the epithelial surface of the respiratory tract, the oviducts and the ependymal lining of the brain ventricles and cerebral aqueducts. Single nonmotile, primary cilia are present on the surface of most cells in the body and have recently been found to play a critical role in sensing the extracellular environment and in the transduction of signalling cascades [10]. They differ from motile cilia in that they lack a central pair of microtubules and dynein motors. The discovery of defects in nonmotile cilia has helped to explain the very diverse clinical features seen in patients with diseases such as polycystic liver and kidney diseases, nephronophthisis, Joubert, Bardet–Biedl, Alstrom, Meckel–Gruber and oral–facial–digital syndromes, Caroli syndrome, and some forms of retinal degeneration. These diseases are now referred to as ciliopathies [11]. In this review we will focus on motile cilia and their structure and function. For further information about other cilia types and ciliopathies we refer readers to some of the excellent recent reviews [11–13].
Motile cilia structure and function
Each motile cilium (fig. 1) is composed of nine peripheral microtubular doublets, interconnected by nexin links, and two central microtubules. Central pair microtubules are linked to the peripheral doublets by radial spokes. Dynein arms are complex protein structures located on the outer and inner side of the peripheral doublets that provide the motor components of the cilia. The outer dynein arms mainly contribute to the frequency of ciliary movement, while the inner dynein arms mainly contribute to the way the cilium bends [15]. A lack of dynein arms or shortening of the dynein arms are the most common structural defects seen in PCD [16–18]. However, there are a number of other defects seen on electron microscopy such as ciliary transposition and radial spoke defects, with new abnormalities being described each year [19]. In some cases of PCD, such as the defect caused by mutation of DNAH11 where cilia often beat rapidly in a dyskinetic fashion, no obvious ultrastructural abnormality can be detected on electron microscopy. It has been reported by some groups that the percentage of patients with PCD with normal ciliary ultrastructure can be as high as 30% [2], although personal experience suggests this is much lower in our cohort of around 300 diagnosed patients. Fortunately, analysis of ciliary beat pattern and beat frequency by high-speed video imaging allows abnormalities of ciliary function to be identified in PCD patients who have a normal ciliary ultrastructure. However, defects in beat pattern can be quite subtle and in the future other forms of diagnostic testing, including immunofluorescence and genetic testing, may be more widely available and provide additional information to aid diagnosis.
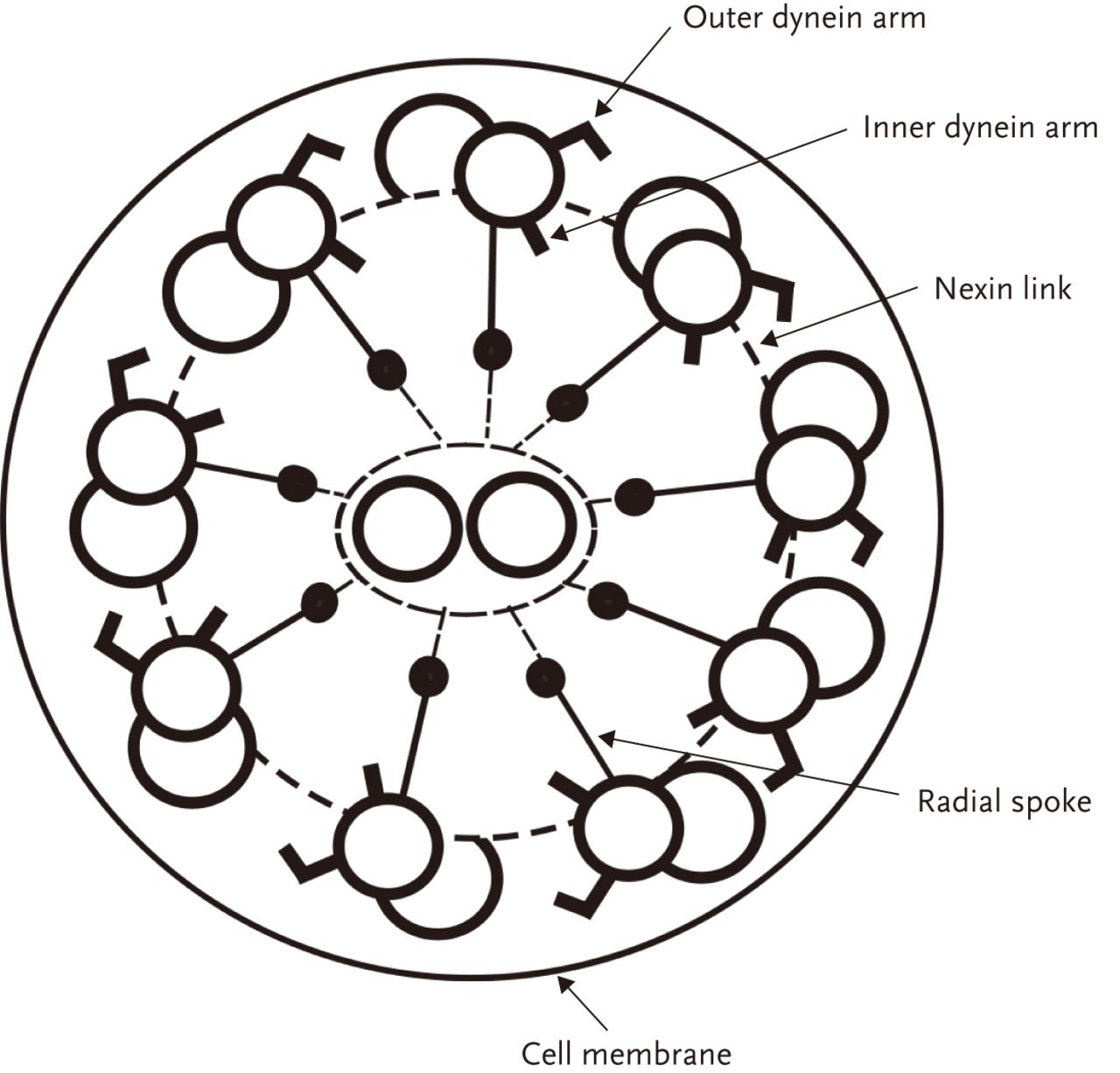
A cross section of the ciliary axoneme. Reproduced from [14] with permission from the publisher.
Pathogenesis of PCD
Mucociliary clearance is one of the main nonimmunological mechanisms that protect us from the pathogens and particles we inhale. Millions of cilia line the surface of the respiratory epithelium and beat in a coordinated fashion, at a frequency of approximately 10–15 Hz, transporting mucus towards the nasopharynx [20]. It is the lack of effective mucociliary clearance in PCD that is responsible for the main clinical problems of recurrent chronic airway infection leading to bronchiectasis, chronic rhinosinusitis and, in many patients, glue ear and associated hearing problems.
Subfertility or infertility in male PCD patients is caused by sperm dysmotility as the sperm tail is structurally very similar to respiratory cilia and powered by dynein motors. Females with PCD may also experience fertility problems due to dyskinetic cilia in their oviducts [21]. Abnormal ciliary function of brain ependymal cilia causes hydrocephalus in small animals with PCD [22]. However, although mild fetal ventriculomegaly may be seen on prenatal ultrasound scans in patients with PCD [23] hydrocephalus is very uncommon in humans [24, 25], except in those with ciliary aplasia (personal observation).
The role of motile nodal cilia in embryonic development has been shown to be crucial for the development of normal situs. Single nodal cilia (9+0), which grow from cells in the primitive node during embryogenesis, lack a central microtubular pair resulting in a circular/oval beat pattern. This produces a unidirectional “nodal flow” that is essential for left–right patterning. If nodal cilia are not moving correctly during this period randomisation of the body axis and organ position occurs resulting in either situs solitus (normal organ position), partial or total situs inversus (inverted position of some or all internal organs) or situs viscerum ambiguus (different forms of heterotaxy) [26–28]. It is important that PCD should be suspected in patients with congenital heart disease who have persistent respiratory and nasal symptoms [28, 29] and particularly in those who have heterotaxy [30].
PCD may also be associated with other conditions including ciliopathies, such as retinitis pigmentosa [31] and polycystic liver or kidney disease [32].
Emerging spectrum of causative genes
At the time of writing, mutations in 26 genes have been shown to cause PCD with a number of others currently undergoing validation [2]. Indeed PCD could be considered as a group of diseases with a remarkably similar phenotype rather than a single disorder.
The gene known to be mutated most commonly in PCD patients is DNAH5 and mutations in this gene may account for 15–30% of all PCD cases [33–35]. Most of the mutations cluster in only a few exons of this huge gene [34, 36]. The protein product of DNAH5 is one of structural proteins of the outer dynein arms. The other known genes known to cause PCD and their corresponding phenotypes are shown in table 1.
Clinical signs and symptoms
Here we highlight some of the important early warning signs and symptoms of PCD that should make clinicians suspect this disorder. This is important, as the age of diagnosis remains worryingly high across Europe [37].
Prenatal and perinatal period
Abnormal organ situs can be detected by ultrasonography during fetal development. Approximately 45% of children with PCD have situs inversus and it has been recommended that children with abnormal organ situs should be referred to a PCD diagnostic centre regardless of other symptoms [38].
A large proportion of, but by no means all, patients with PCD have respiratory problems following birth [39]. This usually presents as unexplained respiratory distress in an otherwise healthy full term baby [2, 40] and a diagnosis of neonatal pneumonia or transient tachypnoea may be made [36]. Classically, a persistent, wet sounding cough is present in the neonatal period and rhinorrhoea/nasal congestion is usually noted by parents [2].
Infants and children
The cardinal sign of PCD is a persistent, wet sounding cough that “has always been there”, never completely clears and is often reported to be worse in the morning. Patients suffer from intermittent or chronic lower respiratory tract infections that ultimately lead to bronchiectasis. Worryingly bronchiectasis has been reported in children under the age of 3 years and is almost universal in adults with PCD [41]. A progressive reduction in lung function is commonly seen [42].
The chronic rhinosinusitis and glue ear seen in children with PCD may result in ear, nose and throat (ENT) referral, but the associated wet sounding cough is often ignored and the diagnosis delayed [43]. Chronic or recurrent otitis media with effusion is very frequent in PCD patients [44], with significant hearing defects present in over half of children [45, 46]. Many children have had multiple ventilation tube insertions before the PCD diagnosis is suspected, which are frequently associated with persistent otorrhoea and in some cases persistent perforation of the ear drum [47]. In Europe hearing aids are advocated, as opposed to surgery, with hearing defects often improving considerably during early teenage years.
It is not uncommon for children to be labelled as having difficult-to-treat asthma, chronic bronchitis, aspiration or an unidentified immune defect [39]. Such a label often defers the diagnosis of PCD. However, despite treatment their daily wet sounding cough and rhinosinusitis never completely clears.
In summary, PCD should be considered in children with situs abnormalities, in term infants with unexplained respiratory distress and in children with a history of a persistent, wet sounding cough for as long as their parents can remember that is associated with persistent rhinosinusitis. It is essential that ENT specialists consider PCD as a diagnosis if a child they review with glue ear and rhinosinusitis has a history of a persistent, wet sounding cough and that cardiologists are aware of the association between heterotaxy and PCD [2].
Adults
A similar spectrum of symptoms is seen in adults although hearing related problems are less common. Lung function decreases with age and it is reported that almost all adults with PCD have bronchiectasis. Noone et al. [41] report that up to 25% of adult PCD patients in the USA are in respiratory failure, with a number requiring lung transplantation [48].
In males, reduced sperm motility (asthenozoospermia) markedly reduces fertility [49]; however, there are reports of successful intracytoplasmic sperm injection [50, 51]. Interestingly, mutations in dynein genes have been reported in male patients with non-syndromic asthenozoospermia without any respiratory problems [52].
Ciliary motility is thought to aid oocyte and early embryo transport through the oviduct, but is not the only mechanism responsible for this [53]. Cilia in the fallopian tubes are also affected in PCD and there are reports of fertility problems in females [21, 54]. It is also believed that defective ciliary activity in the fallopian tubes is responsible for the somewhat higher incidence of ectopic pregnancy seen in patients with PCD compared with the normal population [55].
Diagnosing PCD
Diagnosis can be difficult. For example, some PCD phenotypes show only slight differences in the cilia beat pattern compared with normal cilia and some PCD phenotypes have no detectable ultrastructural defects on electron microscopy [56]. It is, therefore, considered essential that diagnostic testing for PCD should be performed in appropriately equipped centres by staff experienced in the diagnostic methods being used [38]. Video methods that do not allow slow motion replay of beating cilia to assess ciliary beat pattern and the saccharine test are now considered obsolete [38].
High-speed video microscopy
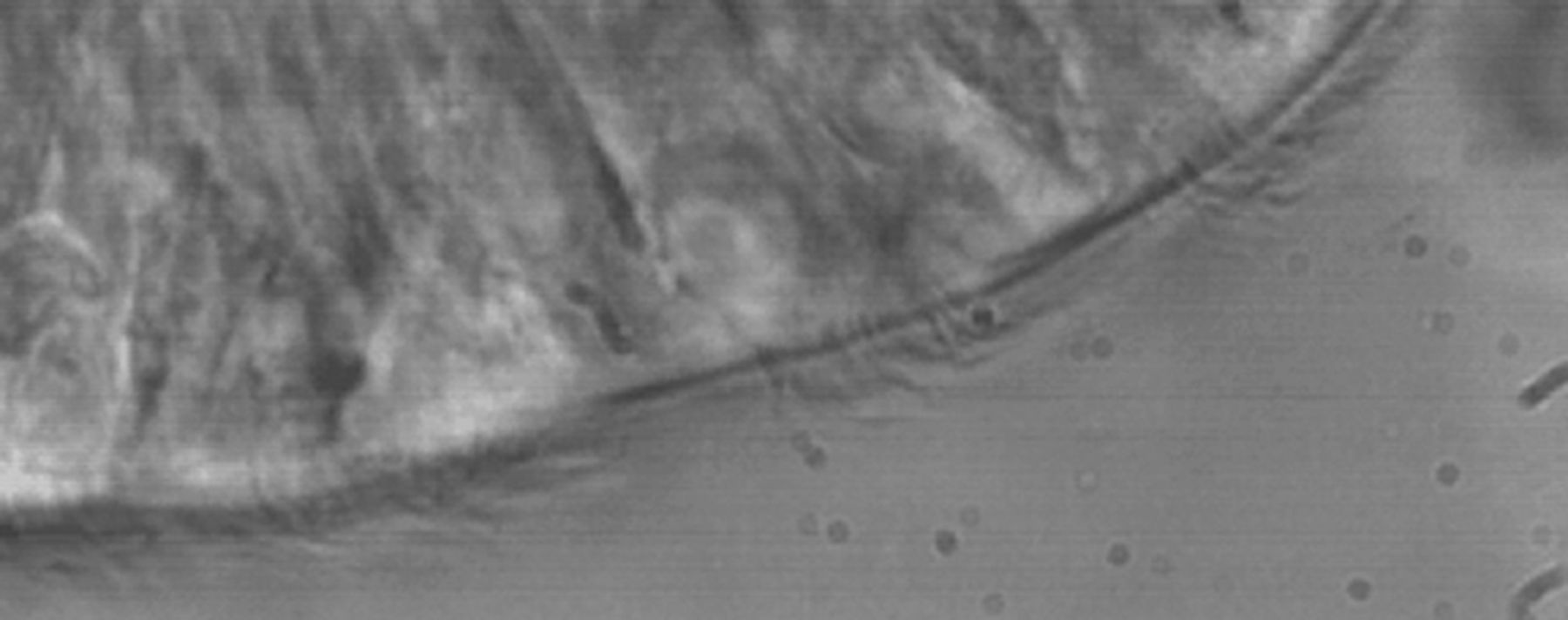
The different ultrastructural defects seen in PCD have been shown to be associated with the abnormalities in ciliary beat pattern observed using digital high-speed video microscopy (fig. 2) [57]. This method has now been adopted as the gold standard for the assessment of ciliary beat pattern [20]. A single nasal brushing can be performed from as early as the first day of life and provides ciliated epithelial strips for high-speed video analysis, electron microscopy and, when required, cell culture. Nasal brushing is quick and easy to perform using a bronchoscopy brush and is well tolerated by patients. Inspection of the nasal passage is essential prior to brushing to exclude structural abnormalities and to identify possible bleeding points. The epithelial edges obtained are observed using a ×100 oil immersion lens and the movement of the cilia is recorded at 500 frames per second. When possible, ciliary beat pattern is observed, in slow motion, on several undisrupted epithelial edges [58] and ciliary beat frequency is measured. The beat pattern of certain PCD phenotypes can be hard to distinguish from the normal ciliary beat pattern and training of staff in beat pattern analysis is essential. Skill is also required to recognise secondary ciliary dyskinesia and to differentiate it from a primary ciliary defect. Observation of widespread secondary ciliary dyskinesia is not infrequent, especially if brushings have been taken within 4 weeks of an upper respiratory tract infection [59–61] and sampling should be avoided during this time.

A single frame taken from a high-speed video recording of a healthy ciliated edge.
Ciliated cell culture can help to confirm the diagnosis in less common PCD phenotypes. Cell culture is also useful when very high levels of ciliary dyskinesia are seen in the sample taken from the patient making interpretation difficult. Following culture a reduction in ciliary dyskinesia is usually seen that reduces the need for repeat biopsy [62].
Transmission electron microscopy
Transmission electron microscopy remains a very valuable diagnostic test and is performed in cases when PCD is suspected from history and high-speed video analysis. However, it can no longer be regarded as the gold standard test for PCD as it is now recognised that PCD may be present where no detectable ultrastructural abnormality can be found [2, 5]. It has been reported that the proportion of patients without an identifiable structural defect of cilia can be as high as 30% [2], although a much lower percentage is seen in our diagnostic centres. Common ultrastructural defects in PCD include absence or shortening of either the inner or the outer or both dynein arms. An increasing number of other defects have been recognised including microtubule disorganisation or transposition (with central pair defects), nexin link defects, central microtubular agenesis and ciliary aplasia [16–19, 63]. Ciliary disorientation, where cilia in the same cell point in different directions, was previously thought to be a common cause of PCD [64, 65]. However, when brush samples are taken at least 1 month after an upper respiratory tract infection this is rarely seen suggesting it is a secondary defect in the vast majority of cases [66].
Nasal nitric oxide
Nasal nitric oxide is an excellent screening test for patients suspected of PCD [67] and several methods are available to measure nasal nitric oxide [68, 69]. The vast majority of patients with PCD have very low nasal nitric oxide values. The reason for this remains unclear [70]. However, it is recognised that a small number of patients with PCD have a normal nasal nitric oxide and that false positives may occur in conditions such as cystic fibrosis (CF) and nasal polyposis [71]. A degree of coordination is required for the test that most children over the age of 5 years can achieve [72].
Other diagnostic tools: cell cultures, immunofluorescence staining and genetic analysis
Ciliated cell culture is particularly useful in patients with persistent widespread secondary ciliary dyskinesia, as it reduces the need for repeat biopsy [62] and aids the diagnosis of rare PCD phenotypes [73, 74]. Following ciliated cell culture, secondary ciliary dyskinesia is reduced compared with that seen in cells taken from the initial biopsy [75] and in PCD the abnormality in beat pattern is often accentuated [76].
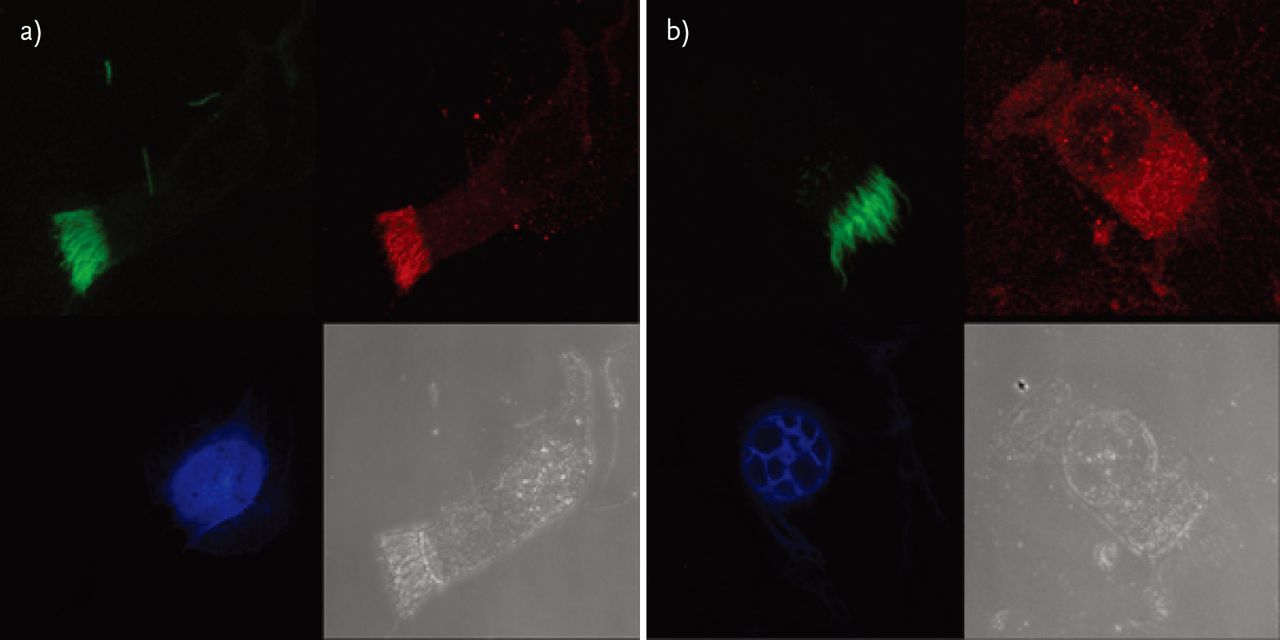
Immunofluorescence staining is based on the knowledge that structural cilia proteins are mislocalised in some PCD patients [77]. Labelling such proteins with fluorescent antibodies may be used for research, as well as for diagnostic purposes (fig. 3).

{kind=link}
{kind=link}
{kind=link}
Immunofluorescence staining of respiratory cells. In each panel the cell is seen in the bottom right-hand corner. The other three images represent the same cell seen in the channel for each immunofluorescence colour. Cells were immunolabelled as follows. Green: anti-acetylated tubulin antibody; blue: DAPI (the nucleus staining); red: anti-DNAH5 antibody. a) In a healthy individual the DNAH5 protein is clearly detectable in cilia (compare the red and the green channel). b) In a patient with PCD caused by mutations in DNAH5 gene the resulting altered DNAH5 protein is mislocalised and detectable only in the cell cytoplasm but not in the cilia (compare the red and the green channel).
At present it is estimated that causative genes can be identified in about 65% of PCD patients [2]. With the decreasing costs of novel genetic tools, genetic testing using next generation and whole genome sequencing will become increasingly available for diagnostic and genetic counselling purposes.
Management of PCD patients
Although research is in progress to restore ciliary function using gene [78, 79] and small molecule therapy, it is unlikely that such interventions will be developed for clinical use in the near future. However, it is generally believed that early diagnosis followed by good respiratory care may prevent or reduce the development of bronchiectasis [80]. Significant variations in management strategies for PCD are seen across Europe [81] and the European Respiratory Society Task Force [38] acknowledges that treatment recommendations are currently based on a very low level of evidence or simply extrapolated from CF guidelines. As the pathophysiology of CF and PCD are fundamentally different such extrapolation is unsatisfactory.
Respiratory management is currently focused on enhancing airway clearance, with a combination of physiotherapy and physical exercise, and aggressive treatment of respiratory infections [38]. As with CF it is felt that patient outcome will be improved if treatment is conducted at a centre experienced in the care of PCD patients.
Culture of sputum or cough swabs is recommended on a regular basis. Haemophilus influenzae, Streptococcus pneumoniae and Staphylococcus aureus are frequently found, with Pseudomonas aeruginosa and nontuberculous mycobacteria seen less frequently. Bronchoalveolar lavage may be required to obtain specimens when sputum is not forthcoming. Oral antibiotics are recommended when there is a worsening of respiratory symptoms or deterioration in lung function, based on culture results where possible. Prophylactic antibiotics are used in some centres, but again there is no evidence to support their use. European guidelines suggest intravenous antibiotics should be considered to treat children not responding to oral antibiotics and if P. aeruginosa is isolated in an attempt to eradicate it. For established P. aeruginosa infection long-term nebulised anti-pseudomonal antibiotics are suggested.
Other therapies including bronchodilators, recombinant human DNase and hypertonic saline have no evidence to support their use. It is of interest that, as with CF, spontaneously expectorated sputum reveals predominantly neutrophilic inflammation [82]. Evidence is awaited to support the use of anti-inflammatory agents such as corticosteroids and azithromycin.
Spirometry should be performed on a regular basis as abnormal lung function can develop early in life and worsens with increasing age. Chest radiographs are insensitive at identifying bronchiectasis when compared with a computed tomography scan, but the latter should be considered carefully to avoid excessive radiation [83].
Childhood immunisations are advocated for all patients as well as pneumococcal immunisation and yearly influenza vaccination [38]. The growth of children should be routinely checked as growth retardation may develop [84].
Various physiotherapy techniques are advised in PCD, but there is no evidence to support the efficacy of any one technique over another. Exercise is strongly advocated for all ages and has been shown to be better than a bronchodilator therapy in PCD [85].
ENT problems in PCD
In children affected by PCD, the respiratory epithelium in the nasopharynx, middle ear and paranasal sinuses is affected resulting in ear and nose problems. Nasal and ear problems associated with a persistent, wet cough should prompt ENT specialists to consider PCD.
Around 85% of the children with PCD have chronic and sometimes severe otitis media with effusion (glue ear). Fortunately, hearing problems associated with this usually improve spontaneously by early teenage years [45]. However, hearing loss and otological problems may also be seen in adulthood [86]. The evidence on management of otitis media with effusion in PCD patients is inconclusive [87]. In Europe, otitis media with effusion is usually managed conservatively with hearing aids, as treatment with ventilation tubes (grommets) often results in prolonged and offensive otorrhoea [88, 89].
Increased nasal secretions are almost universal in children with PCD with parents frequently describing their child as having a permanent cold [90]. Nasal polyps are reported in up to 18% of patients [91]. Saline nasal douches and anti-cholinergic therapy are often used to treat symptoms, but again there is no evidence base to support their use. It has been suggested that some patients with chronic rhinosinusitis may benefit from long-term macrolide therapy and from endoscopic sinus surgery in recalcitrant disease [86].
Conclusions
Raising awareness of the diagnosis of PCD is essential as there is evidence that appropriate respiratory care may reduce the decline in lung function. It is a matter of concern that the diagnosis of PCD is often made late, even in children with situs inversus.
Diagnostic testing for PCD has improved over recent years and should be available in specialist centres in each country. New genotypes of PCD are being discovered each year and it is likely many more will be discovered.
Research is urgently needed to provide evidence-based treatment for patients with PCD and the development of cohorts of patients with PCD will allow research studies to proceed.
Educational questions
-
PCD is:
-
A polygenic inherited disease
-
A genetically heterogeneous condition
-
Usually autosomal recessive
-
Usually diagnosed very early in childhood
-
-
Most PCD patients:
-
Have neonatal respiratory distress syndrome and low birth weigh
-
Have organ situs abnormalities
-
Have a history of otitis media with effusion
-
Have only a functional defect of cilia (no structural defect of cilia can be detected)
-
-
When diagnosing PCD:
-
More than one diagnostic test is usually needed
-
Genetic testing is always required to confirm the diagnosis
-
Exhaled nitric oxide is often used
-
Ciliary movement is assessed using high-speed video microscopy
-
-
Cilia:
-
Can be found on the majority of human cells
-
Developed late during evolution
-
Beat in a coordinated manner on the respiratory epithelium
-
Participate in signal transduction in the kidneys
-
-
Considering the management of PCD patients:
-
Bronchodilators are regularly used to treat the bronchial obstruction
-
Regular respiratory physiotherapy and exercise are the main therapeutic procedures
-
Regular audiograms do not have to be performed in adult patients
-
Chest radiographs are recommended to monitor the progress of the lung disease
-
Suggested answers
-
b, c.
-
c.
-
a, d.
-
a, c, d.
-
b.
Footnotes
The ERS designates this educational activity for a maximum of 1 CME credit. For information on how to earn CME credits, please visit www.ers-education.org/e-learning/cme-tests
Statement of Interest
None declared.
- ©ERS 2014
Breathe articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










