





Abstract
Educational aims
To recognise the clinical and radiological presentation of the spectrum of diseases associated with bronchiectasis.
To understand variation in the aetiology, microbiology and burden of bronchiectasis and cystic fibrosis across different global healthcare systems.
Bronchiectasis is the term used to refer to dilatation of the bronchi that is usually permanent and is associated with a clinical syndrome of cough, sputum production and recurrent respiratory infections. It can be caused by a range of inherited and acquired disorders, or may be idiopathic in nature. The most well recognised inherited disorder in Western countries is cystic fibrosis (CF), an autosomal recessive condition that leads to progressive bronchiectasis, bacterial infection and premature mortality.
Both bronchiectasis due to CF and bronchiectasis due to other conditions are placing an increasing burden on healthcare systems internationally. Treatments for CF are becoming more effective leading to more adult patients with complex healthcare needs. Bronchiectasis not due to CF is becoming increasingly recognised, particularly in the elderly population. Recognition is important and can lead to identification of the underlying cause, appropriate treatment and improved quality of life. The disease is highly diverse in its presentation, requiring all respiratory physicians to have knowledge of the different “bronchiectasis syndromes”. The most common aetiologies and presenting syndromes vary depending on geography, with nontuberculous mycobacterial disease predominating in some parts of North America, post-infectious and idiopathic disease predominating in Western Europe, and post-tuberculosis bronchiectasis dominating in South Asia and Eastern Europe.
Ongoing global collaborative studies will greatly advance our understanding of the international impact of bronchiectasis and CF.
Abstract
Bronchiectasis and CF: disabling diseases with a growing impact worldwide http://ow.ly/U0S0302ocv3
What is bronchiectasis?
Bronchiectasis is an anatomical description, referring to abnormal widening of the bronchi which may be caused by a wide range of inherited and acquired disorders [1–3].
Radiologically, bronchiectasis is most easily identified by computed tomography (CT) [3]. At any level, the diameter of the bronchi should be less than the adjacent blood vessel (known as the bronchoarterial ratio) [4]. An increase in the diameter of the bronchus, and a failure of the bronchus to taper normally as it goes out towards the periphery of the lung, is diagnostic of bronchiectasis. Other radiological features that may accompany bronchiectasis include bronchial wall thickening, mucus plugging, tree-in-bud abnormalities, air trapping, mosaicism and emphysema [4, 5]. None of these features are required for diagnosis.
Bronchial dilatation is cylindrical when mild, varicose when more severe and cystic when most severe (figure 1) [4–6].

Radiological heterogeneity of bronchiectasis. Two patients are shown: a) a patient with moderate idiopathic bronchiectasis demonstrating predominantly cylindrical dilatation, and b) a patient with severe disease and Pseudomonas aeruginosa colonisation demonstrating widespread varicose and cystic bronchial dilatation.
When the term of bronchiectasis is used to refer to a disease rather than a radiological appearance, it describes a clinical syndrome of cough, sputum production and recurrent respiratory tract infections [1, 2, 7]. Dilatation of the bronchi leads to failure of normal mucociliary clearance, which may be exacerbated by abnormally thick mucus such as in cystic fibrosis (CF) [8–10]. The airways become colonised with bacteria, which promotes recruitment of neutrophils and other inflammatory cells that further damage the airway through complex mechanisms that include the release of cytokines/chemokines, proteases and antimicrobial peptides that combine to promote mucus hypersecretion, failure of bacterial killing, airway remodelling and failure of ciliary function [10–14]. This establishes “the vicious cycle of bronchiectasis”, which has been well described [13]. Whether failure of mucociliary clearance, inflammation or infection occurs first to initiate the cycle is unclear, and it is likely the initiating event is different depending on the underlying cause of bronchiectasis [14–16].
What causes bronchiectasis?
The majority of cases of bronchiectasis are not due to CF, so-called non-CF bronchiectasis [17]. Bronchiectasis therefore represents a final common pathway of a large number of disorders [15–19].
Post-infectious bronchiectasis occurs when bronchiectasis develops in adults or children following a severe infection [19]. This may arise following pneumonia, and is described frequently following infection with Mycobacterium tuberculosis. Nontuberculous mycobacteria (NTM), such as M. avium and M. abscessus, are strongly associated with bronchiectasis and CF, although in the case of non-CF bronchiectasis is not always clear whether NTM is cause or a complication of bronchiectasis [20, 21]. Viral infections are also described as a cause of bronchiectasis [22]. Bronchiectasis is an important complication of primary immunodeficiencies such as common variable immunodeficiency, where it is presumed that recurrent infections lead to progressive lung damage [23]. It is important to be aware that, although typically presenting in children, immunodeficiencies may be first diagnosed in adulthood.
A large number of connective tissue diseases (CTDs) have been associated with bronchiectasis [24, 25]. The association between rheumatoid arthritis and bronchiectasis is well described, with 2–10% of rheumatoid arthritis patients being affected. Sjögren’s syndrome, systemic lupus erythematosus, ankylosing spondylitis, scleroderma and vasculitis have all been reported to have a higher incidence of bronchiectasis than that expected in the general population [24]. In many cases associated with CTD, the patient may be entirely asymptomatic. The significance of abnormal bronchial dilatation in the absence of symptoms in patients with CTD is unclear.
Inflammatory bowel diseases (IBD), both ulcerative colitis and Crohn’s disease are associated with bronchiectasis [26]. Airway involvement in IBD is thought to be rare (reported at ∼0.2% in an IBD clinic) and progresses through chronic bronchitis, without radiological bronchiectasis, through inflammatory bronchorrhea (which can be very large volume, e.g. >100 mL⋅day−1) to classical bronchiectasis [26].
Allergic bronchopulmonary aspergillosis (ABPA) is an important cause of bronchiectasis to identify, because it has a specific treatment that can improve symptoms and prevent the development of severe bronchiectasis [27, 28]. Patients typically have a history of asthma (or CF) and present with cough and sputum (often with thick plugs and large volumes) with deteriorating symptoms of asthma including wheeze and exacerbations. Patients may have fleeting infiltrates on chest radiogrpahy with segmental, lobar or lung collapse. The bronchiectasis is typically proximal, but this is not universal [1]. Diagnosis depends on demonstrating a specific hypersensitivity response to Aspergillus fumigatus (or occasionally other fungi) on laboratory testing.
Inhaled foreign bodies, lung tumours and other obstructive lesions can cause localised bronchiectasis, and consequently bronchoscopy is advised where bronchiectasis affects a single lobe [1].
There are a number of congenital or inherited causes of bronchiectasis, most of which are rare.
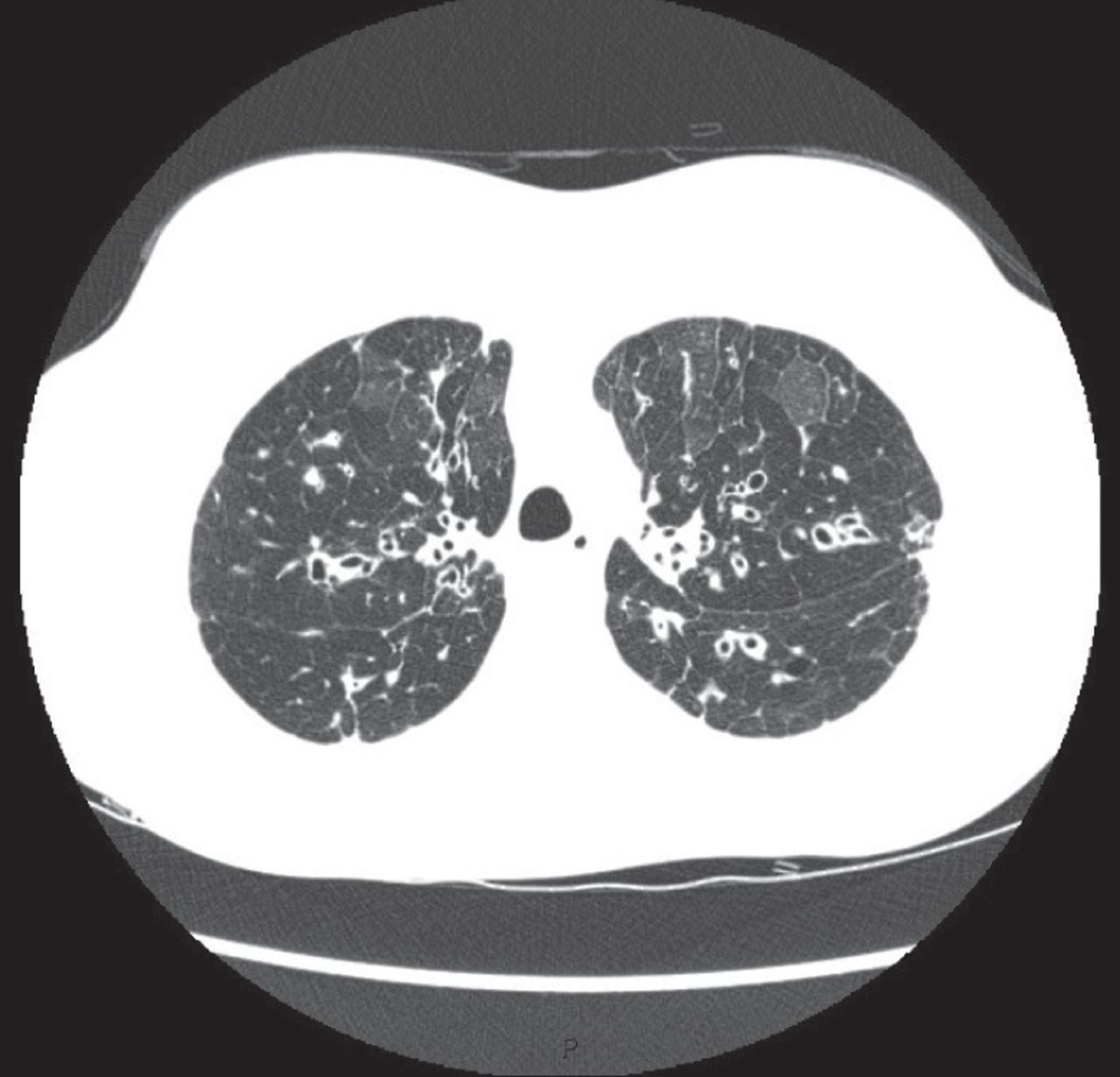
CF is an autosomal recessive condition affecting 1–10 per 100 000 people across Europe. One in 25 Caucasians are carriers of mutations known to cause CF [29]. The carrier frequency in other ethnic groups is lower and consequently the prevalence of the disease is lower. It is a multisystem disorder resulting from mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) [30]. The majority of cases are now detected through newborn screening in Western countries, although diagnosis in adulthood in cases initially thought to be “non-CF” bronchiectasis is not uncommon [1, 30]. CF should be excluded in all children presenting with bronchiectasis and in adults presenting under the age of 40 years, in those with upper lobe bronchiectasis (figure 2), in those with extrapulmonary manifestations (malabsorption, pancreatitis or intestinal obstruction), and in those with Staphylococcus aureus, Pseudomonas aeruginosa or NTM [1, 2, 9].

High-resolution CT scanning in a 27 year old patient with homozygous ΔF508 CF. Bilateral upper lobe bronchiectasis is evident, with mosaicism. The scan also showed extensive emphysema.
CF has a special importance, because of the huge morbidity associated with the disease, which despite great advances in its treatment still has a life expectancy of only 40–50 years [31]. When it was first recognised as a distinct disease in 1938, patients with CF were expected to live only for a matter of months [32]. As antibiotic therapy and pancreatic enzyme replacement were developed in the mid-20th century, life expectancy and quality of life improved; however, these patients continued to die in adolescence or early adulthood. The past three decades of international translational research and refinement in the practice of caring for patients with CF, however, has resulted in further advances in nutritional therapy, targeted inhaled anti-pseudomonal agents and airway clearance, and most recently CFTR modulators that open the possibility of correcting the basic pathophysiological mechanism of CF [33]. The cumulative effect of such research and development is that many patients with CF can now expect to take on the tasks of everyday adult life such as finishing school, joining the workforce and starting families. CF is now a model of how specialist care, translational research and interaction between healthcare providers, pharmaceutical industry and charities can drive improvements in life expectancy and quality of life for patients [8, 29–33].
Primary ciliary dyskinesia (PCD) is also an autosomal recessive disease [34]. The prevalence of the disease is unknown and likely to be greatly underestimated due to the absence of widely available screening tests. The disease may be diagnosed in childhood, or may be identified in late adulthood, with of cases reported beyond the sixth decade of life [35, 36]. The classical presentation of PCD includes childhood onset of respiratory symptoms, recurrent otitis media and sinusitis. Approximately half of patients with this classic presentation will have dextrocardia [35]. Other forms of congenital abnormalities may be present such as complex congenital heart disease, oesophageal atresia, biliary atresia or hydrocephalus. Infertility may be present. Young’s syndrome is a rare subset of PCD defining the combination of bronchiectasis, rhinosinusitis and reduced fertility (azoospermia).
α1-antitrypsin deficiency (AATD) lung disease is typically characterised by airflow obstruction, but a significant number of patients have bronchiectasis [37, 38]. ∼40% of patients have chronic bronchitis and sputum production [37]. In one series from the UK of 74 subjects with PiZ AATD, 20 had radiological and clinically significant bronchiectasis [38]. Since bronchiectasis is typically not the only feature of AATD, screening for AATD in unselected patients with bronchiectasis has limited value [39]. Nevertheless, AATD should be suspected in patients presenting at a young age with prominent emphysema and airflow obstruction, particularly in the presence of basal emphysema on CT [1].
Myeloma and haematological malignancy are rarely described in association with bronchiectasis, but there is a perception that this group of patients are becoming more prevalent. McShane et al. [40] studied 16 patients with haematological malignancy in a US series. Cases associated with bronchiectasis included acute lymphocytic leukaemia, chronic lymphocytic leukaemia, acute monocyte leukaemia, chronic monocytic leukaemia, non-Hodgkin lymphoma and Waldenstrom’s macroglobulinaemia. Graft versus host disease following stem cell transplantation is a recognised cause of bronchiectasis, but in this series nearly half of patients had not received stem cell transplantation and so it is likely defective immune function directly related to the haematological malignancy contributes to the development of bronchiectasis [40, 41]. All patients with bronchiectasis should be screened with a full blood count and measurement of serum immunoglobulins, which may detect primary and secondary immunodeficiency. Serum electrophoresis and Bence Jones protein in urine are used to screen for myeloma [1].
Gastro-oesophageal reflux disease (GORD) and aspiration are reported to be causes of bronchiectasis, although this association has been poorly studied and it is difficult to ascertain to what extent GORD is a genuine aetiology because it is so prevalent in the general population. Some data suggests GORD may be a disease modifier more than an aetiology [42, 43].
HIV infection may predispose to bronchiectasis through susceptibility to recurrent infections [44]. There is also a strong link between HIV infection and tuberculosis (TB) or NTM infection, both of which are strongly linked to bronchiectasis [44, 45]. In the era of highly active anti-retroviral therapy, opportunistic infections are less common but the impact of HIV on the immune response is also highly relevant. HIV patients have a higher prevalence of smoking that the general population and are more vulnerable to smoking-related lung damage and the development of chronic obstructive pulmonary disease (COPD) [45]. Therefore, in addition to being more susceptible to a number of disorders associated with bronchiectasis, even in the absence of these, patients with HIV appear to have a higher incidence of bronchiectasis than expected in the general population [46].
Yellow nail syndrome is a very rare orphan disease characterised by the triad of yellow nails, chronic respiratory disease and primary lymphedema (often presenting as pleural effusion). Males and females are equally affected and patients will usually develop symptoms in late childhood/early adulthood, although diagnosis may be delayed until very late in life. The triad is not always present, as nail changes can occur without respiratory involvement, and similarly lymphedema may be absent [47].
Mounier–Kuhn syndrome (also known as tracheobronchomegaly) is a congenital disorder characterised by marked dilation of the trachea and proximal bronchiectasis. It is more frequent in men and often presents in early adulthood with typical symptoms of bronchiectasis, including recurrent infections, cough and sputum production. It occurs due to atrophy of the elastic fibres of the trachea and main bronchi leading to tracheobronchial flaccidity, dilatation and collapse. It is likely to have a genetic basis but a causative gene has yet to be identified (figure 3) [48].

Two images from the same individual with tracheobronchomegaly showing a) gross tracheal dilatation and b) severe central bronchiectasis.
Radiological bronchiectasis without symptoms should be differentiated from clinically significant bronchiectasis. Some studies report a very high rate of bronchiectasis in healthy nonsmokers. For example, Winter et al. [49] conducted CT scans in 47 healthy persons older than 65 years and 24 persons aged 30–50 years, and reported bronchiectasis in 14.9% of those aged >65 years. In the oldest patients aged <75 years, 19.1% had bronchiectasis. Matsuoka et al. [4] performed high-resolution CT in 85 patients without known respiratory disease and reported the presence of bronchiectasis (defined as a bronchoarterial ratio >1) in 41% of subjects older than 65 years. Since none of these patients had symptoms, this suggests a very high frequency of asymptomatic mild bronchial dilatation in the elderly [4]. Bronchoarterial ratio increased with age and smoking also appeared to increase the ratio in the elderly population [4].
This list of causes is not exhaustive, but outlines both the most frequent causes of bronchiectasis in children and adults while also emphasising the highly variable clinical presentation of these diseases (table 1). Despite extensive clinical, radiological and immunological characterisation, as recommended by consensus guidelines, 30–70% of patients are classified as idiopathic bronchiectasis following testing [2–5].
Selected causes of bronchiectasis with their reported prevalence
What are the links with “traditional” airway diseases (COPD and asthma)?
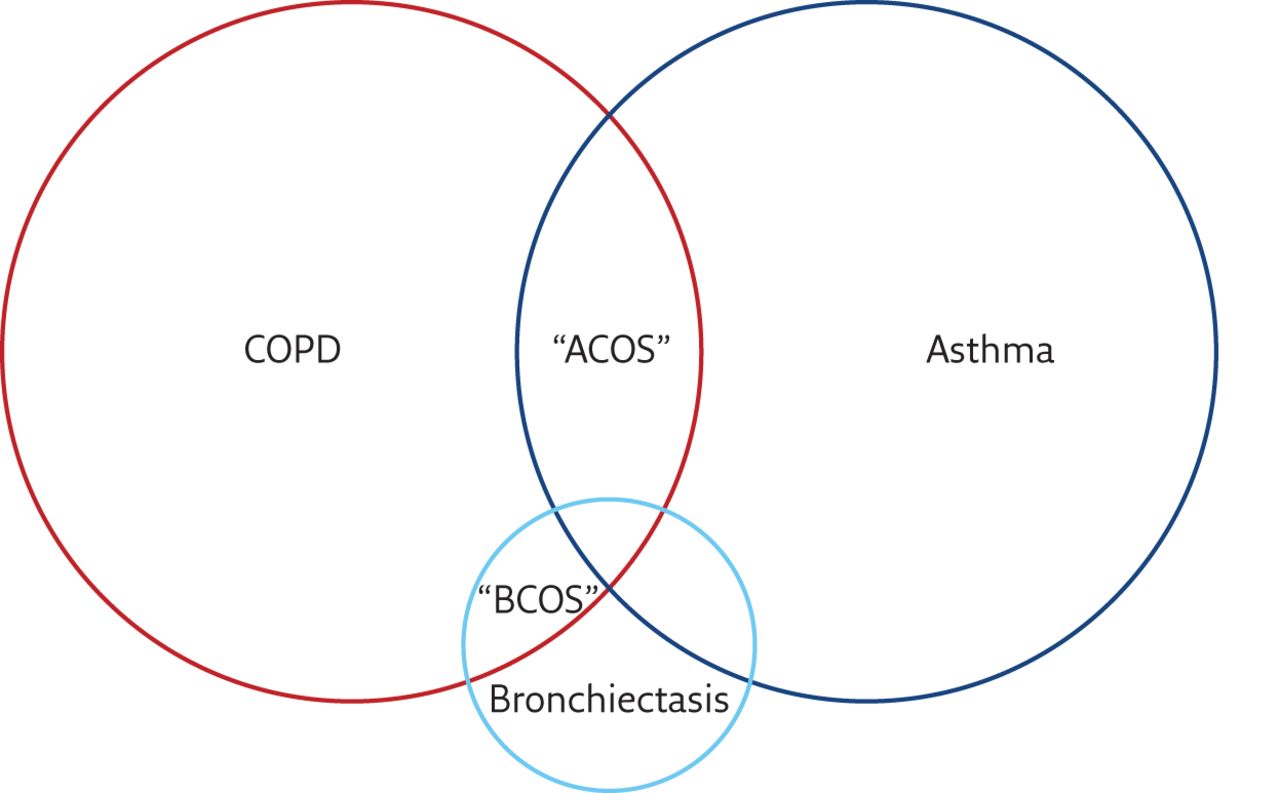
There is overlap between COPD and asthma (sometimes referred to as asthma–COPD overlap syndrome (ACOS)) and there is clearly also overlap between bronchiectasis and COPD (sometimes called bronchiectasis–COPD overlap syndrome (BCOS)) [50–52]. A large proportion of patients with bronchiectasis will be initially diagnosed with COPD or asthma. In the recent series described by Quint et al. [53], using a large primary care database in the UK to study the prevalence of bronchiectasis, 42.5% of patients with bronchiectasis had a coexisting diagnosis of asthma and 36.1% had a coexisting diagnosis of COPD. Whether this represents genuine co-diagnosis, or whether there is a degree of misdiagnosis is unclear. There is clear overlap between these three disorders (figure 4); therefore, a patient presenting to primary care with cough, sputum production and airflow obstruction on spirometry is likely to be initially diagnosed with COPD, particularly if they have a smoking history [54]. Triggers for further investigation may be a failure to respond to standard treatment, frequent exacerbations, large volume sputum production, isolation of Gram-negative organisms in sputum, or severity of COPD that seems out of proportion to the reported smoking history [55]. The same applies to asthma where a high prevalence of bronchiectasis is likely to represent both a degree of misdiagnosis and coexistence of the two conditions at the same time as well as potentially chronic inflammation in asthma leading to the development of bronchiectasis [56, 57].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Venn diagram illustrating the overlap between COPD, asthma and bronchiectasis.
The prevalence of bronchiectasis in COPD patients is highly variable in the literature. The largest series such as the ECLIPSE cohort (n=2164) report a frequency of 5% in Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage III and 7% in GOLD stage IV disease [58]. However, how bronchiectasis was radiologically defined in this cohort is not clear [58]. Another large cohort, COPDgene (n=3636), has reported a prevalence of 20.8% in COPD patients, with an association between radiological bronchiectasis and exacerbations and worse forced expiratory volume in 1 s (FEV1) [59]. While this may imply a high frequency of bronchiectasis, when put into the context of the study by Matsuoka et al. [4] (discussed earlier) suggesting 41% of asymptomatic healthy adults aged >65 years have radiological bronchiectasis, it cannot be said for certain whether COPD patients have a greater frequency of bronchiectasis than the general population [4].
Nevertheless, there is a consistent trend across multiple studies for bronchiectasis to be more frequent as airflow obstruction becomes more severe [60]. It has also been suggested that COPD and bronchiectasis together are associated with greater rates of bacterial colonisation and even death, although this is not a consistent finding and a recent meta-analysis pointed to some publication bias (where smaller studies with large effects were being preferentially published) [60]. Looking at this association from a bronchiectasis perspective, rather than from the point of view of COPD, where a bronchiectasis patient has airflow obstruction or a clinical diagnosis of COPD, there is a significantly increased risk of death demonstrated across multiple studies [61–63].
There are less data available regarding asthma. Paganin et al. [57] performed high-resolution CT scanning on 70 patients with allergic and 56 patients with non-allergic asthma. They demonstrated cylindrical bronchiectasis in up to 80% of patients with severe non-allergic asthma and nearly 50% of patients with severe allergic asthma [57]. In patients with mild disease, the corresponding rates were ∼50 and ∼20%, respectively. The authors concluded that bronchiectasis is common and related to the severity of asthma. Recently, asthma has been proposed as a contributor to exacerbations of bronchiectasis [56]. Again, because asthma is common in the general population, it is difficult to determine to what extent asthma is a cause rather than a coexisting condition. Applying the Bradford–Hill concepts for causality, there remains much work to be done in this area (table 2).
Bradford–Hill criteria for causality applied to the association between COPD, asthma and bronchiectasis
The association between asthma and CF bronchiectasis is similarly unclear, as the overlap in symptomatology and variable airflow obstruction makes diagnosis challenging. Further obfuscating the diagnosis is the recognition of bronchial hyperresponsiveness in CF cohorts and bronchiectasis cohorts [64, 65]. Despite this, many patients with CF and bronchiectasis are treated with inhaled corticosteroids and other typical asthma therapies without well-established evidence of benefit [66]. Further research is needed to establish best practices of care for these patients.
What is the clinical presentation of bronchiectasis?
Bronchiectasis has a highly variable clinical presentation, as it is a disorder that can affect any age from young children to the very elderly, males and females, and it is present in all ethnic groups. As such, it is not possible to describe a single typical clinical presentation. Some examples are discussed below which illustrate the clinical heterogeneity of the condition.
Example 1
A 62-year-old woman, who is a lifelong nonsmoker, presents with a 5-year history of cough and sputum production. The symptoms have become worse recently. She was diagnosed with asthma and given an inhaled corticosteroid, which has had little effect. She improves with antibiotics, but her symptoms recur a few days after stopping antibiotics. Spirometry shows an FEV1 of 88% predicted, with no airflow obstruction. Sputum culture grows Haemophilus influenzae. A high-resolution CT is performed and shows bilateral lower lobe bronchiectasis.
Although it would be important in this case to exclude other possible causes of bronchiectasis, this is the typical presentation of a patient with “idiopathic” bronchiectasis [1]. 60–80% of bronchiectasis patients are female, and presentation is very common between the ages of 50–70 years [62, 63, 67]. The reasons for this preponderance in post-menopausal females are unknown. The delay in diagnosis is common. The good, but short-lived response to antibiotics is also a common feature that should raise suspicion of bronchiectasis [1].
Example 2
A 71-year-old woman, who is a lifelong nonsmoker, presents with a 2-year history of cough, which was largely unproductive, and recent shortness of breath and weight loss. Her current BMI is 18.3 kg⋅m−2. She undergoes a high-resolution CT scan which shows localised bronchiectasis in the right middle lobe with a nodular appearance. She produces two sputum samples that are negative for typical bacterial pathogens and grow no Mycobacteria. Nevertheless, due to clinical suspicion she underwent a bronchoscopy, with the washings isolating M. avium.
A high index of suspicion is required for NTM as it may present in patients without this typical “Lady Windermere syndrome” history. Patients are typically middle aged or elderly females and may have scoliosis and a low BMI [68]. The middle lobe or lingular bronchiectasis and nodular changes are classical, but are not required for diagnosis and so all patients with bronchiectasis should have sputum tested for NTM at diagnosis and during a clinical deterioration. Sputum cultures are sometimes negative for NTM even in those patients with active infection, either due to poor quality samples or low bacterial loads present; therefore, bronchoscopic sampling should be considered in patients with a high index of suspicion, as in this case [68].
Example 3
A 24-year-old man presents with cough and sputum production and persistent rhinosinusitis. His chest radiograph is normal, but a high-resolution CT scan suggests bilateral bronchiectasis. BMI is normal, and there is no suggestion of malabsorption and no history of neonatal distress or respiratory infections. There is no family history of respiratory problems. There is a history of recurrent otitis media during childhood leading to unilateral hearing loss.
This patient has PCD. Although it would be appropriate to exclude CF and immunodeficiency, the history of rhinosinusitis and middle ear infections is typical and these features are almost universal in some series of PCD. Although most cases will be detected in childhood it is important to have a high index of suspicion in patients presenting in early adulthood with bronchiectasis [69].
Clinical impact of bronchiectasis
Bronchiectasis has a major impact on patients in terms of symptoms, exacerbations and quality of life. In severe cases, bronchiectasis is also associated with a life expectancy shorter than that of the general population [70]. In specialist centres, ∼50% of patients will die from respiratory complications such as respiratory failure and respiratory infections, with 50% dying from nonrespiratory causes [62].
Mortality rates have been reported at 2–5% over 2 years, 10–30% at 5 years and up to 35% at 15 years [61, 62, 70].
Patients may experience 0–12 exacerbations per year, with the average in most series being between one and three exacerbation per year [71–73]. Severe exacerbations requiring hospital admission and intravenous antibiotics have a greater impact in terms of healthcare costs, as well as in terms of long-term prognosis as they are associated with more significant lung function decline and increased mortality [62].
The St George’s Respiratory Questionnaire has been extensively used in bronchiectasis as in other respiratory diseases allowing a comparison of the burden of bronchiectasis to other disorders. In this sense, the quality of life impairment seen in bronchiectasis is equivalent to that observed in idiopathic pulmonary fibrosis or moderate-to-severe COPD [12].
The impact of bronchiectasis is highly variable. Mild disease is characterised by the absence of bacterial colonisation, infrequent exacerbations, less radiological extension and fewer symptoms. While features of severe disease include colonisation with P. aeruginosa, frequent and severe exacerbations, severe breathlessness and lung function impairment with more extensive radiological disease [62, 74]. A scoring system incorporating these variables, named the Bronchiectasis Severity Index, has been developed to guide prognostication in bronchiectasis and has been extensively validated in Europe, China and in other healthcare systems [62, 75, 76].
While there is a move towards managing bronchiectasis is specialised clinics or centres in some countries, such as the UK, this is unusual in the majority of countries worldwide. Some patients with more severe bronchiectasis are cared for in CF clinics. A survey by the European Bronchiectasis Network (EMBARC) of 231 European centres suggested that the majority (52%) managed patients in general respiratory clinics [77].
Clinical impact of CF
In the mid-20th century CF was recognised, but was usually fatal in early childhood because of a combination of respiratory infections and nonpulmonary complications such as malabsorption and intestinal obstruction [8, 9]. Progressive improvements in care over time, including the development of pancreatic enzyme replacement, improved nutrition, systemic antibiotic treatment, mucoactive treatments and the recognition and treatment of infection with P. aeruginosa have driven remarkable improvements in life expectancy [8, 9, 78]. Key to these developments has been the establishment of specialised care centres and robust standards of care [79].
CF is now in the process of changing from a predominantly “paediatric” disease to one where the major burden of care is in adults [9]. The median survival reported from the US and other national registries is ∼40 years, and is expected to rise further in the coming years [9].
Despite these improvements there remains a very high burden of disease and treatment associated with CF. Care is highly complex and multisystem including provision for nutrition/dietetics, CF-related diabetes, CF-related liver disease, psychology, physiotherapy and all aspects related to the care of pulmonary complications. Complications of CF in adulthood are most frequently pulmonary, and include severe exacerbations, respiratory failure, pneumothorax, massive haemoptysis and requirement for lung transplantation [8, 9, 79]. CF care requires centralisation and concentration of expertise. The European Cystic Fibrosis Society standards of care describe the optimal make-up, size and facilities available in a specialist CF centre. Specialist CF centres should typically care for more than 100 patients [79].
Bronchiectasis and CF in Europe
Current prevalence estimates for bronchiectasis, excluding CF, are highly variable across different healthcare systems. German data report a rate of 67 per 100 000 while accepting this may be an underestimate [80]. Recent UK data suggests the condition is surprisingly common with rates of prevalence of 566 per 100 000 for women in the most recent year of data (2013). Rates were even higher when limited to the population aged >65 years, approaching 1% of the population [53]. Whether these UK data are applicable more widely across Europe is not known.
Data suggest that there is some variability regarding the aetiologies across Europe. Post-infectious bronchiectasis is the most commonly identified aetiology across European countries, although the data suggest that is more frequent in the southern countries (Greece, Italy and Spain) [16]. A recent analysis from Greece found that the leading cause of disease was post-TB, reflecting the higher rates of TB prevalence in Eastern or South-east Europe compared with Western Europe, from where the bulk of publications on bronchiectasis have arisen to date [81–83].
CF is more prevalent in Europe, North America and Australasia, and occurs mainly in children of European descent [84]. Across the European countries, Ireland has the highest incidence rate, while the UK and Italy have also a high incidence of CF [85]. Eastern Europe (Poland, Bulgaria and Romania) and Baltic countries (Latvia and Lithuania) have a much lower incidence [84, 85].
Bronchiectasis and CF in North America
There are more than 28 000 people in the USA with CF. These patients are cared for in regional, multidisciplinary CF centres accredited by the CF Foundation. Tremendous advances in diagnostics and therapeutics over the past two decades have resulted in a shift in demographics in the USA such that the majority of patients with CF are now >18 years of age [86].
While patients with CF are generally cared for by centres accredited by a well-organised system, the majority of patients with non-CF bronchiectasis in the USA are cared for by pulmonologists in settings varying from general pulmonary practice to dedicated, but not standardised, bronchiectasis clinics. In 2007, the COPD Foundation Bronchiectasis and NTM Research Registry was established to better characterise the burden of disease in the USA and to coordinate research efforts. This collaborative initiative has approximately a dozen participating sites including academic research institutions, which are referral centres for refractory or challenging cases and have particular experience in caring for difficult-to-treat infectious aetiologies, such as NTM [87]. Earlier prevalence estimates of non-CF bronchiectasis in the USA were ∼52 per 100 000, which is close to the European Union definition of an orphan disease (50 per 100 000). More recently, Seitz et al. [88] reported a prevalence in the Medicare outpatient claims database of 370 per 100 000, with the highest prevalence occurring in females aged 80–84 years (prevalence of 537 per 100 000).
The rates of NTM lung disease in patients with non-CF bronchiectasis in the USA have been highly variable, but are reported to range from 10 to 30% with an overall increasing trend [89]. There may be some bias in these rates as data are collected from referral centres with regional expertise in caring for NTM disease. The rates of NTM in CF patients in the USA have been reported to range from 10 to 20% with considerable geographic variation [90]
Bronchiectasis and CF in Asia
Bronchiectasis is a relatively common disease in the Asia-Pacific region. However, there is scant research interest and hence the true prevalence is unknown. The disease is underdiagnosed and many patients are diagnosed as COPD or asthma. The disease is thought to be more prevalent in those of lower socioeconomic status where immunisation rates are low [91, 92]. Bronchiectasis is very common in indigenous Australians and particularly in children [93].
Hong Kong government statistics show a hospital admission rate of ∼16.4 per 100 000, and the mortality rate was 1 per 100 000 population in 1990 [94]. Under-reporting is a major problem because bronchiectasis patients are treated primarily on an outpatient basis.
Idiopathic disease still seems to be the commonest cause of bronchiectasis, as in the West, but there is a much higher rate of post-TB bronchiectasis in some Asian countries. Among 100 patients with bronchiectasis analysed in Hong Kong 82% were found to have idiopathic disease and 8% had post-TB bronchiectasis [95]. The low rate of post-TB bronchiectasis reflects the effective TB control programme in Hong Kong. Preliminary data from the Indian bronchiectasis registry (EMBARC India; unpublished data) also shows that idiopathic disease is the most common cause, even though the incidence of post-TB bronchiectasis is much higher in India compared with Hong Kong. Epidemiological data from Thailand shows that post-TB bronchiectasis is the most common cause in that country [96]. Studies emerging from Hong Kong, Busan in South Korea and EMBARC India show that P. aeruginosa is the commonest organism infecting the airways of patients with bronchiectasis [97]. M. avium complex pulmonary disease with nodules and bronchiectasis is also increasingly recognised in Asia including Japan [98]. NTM, which included M. kansasii and M. chelonae, were found in 6% of cases from Thailand [96].
Classical CF, with pancreatic insufficiency and progressive lung disease, is rare in the non-white population despite the greater presence of F508 mutation among the indigenous South Asian population [99]. The prevalence of classical CF in Japan is 1 in 350 000, while sporadic cases have been reported from indigenous Sri Lankans, Indians, Pakistanis, Chinese, American-Indians, black Africans, Malays and Arabs [100, 101].
Management
Treatment of bronchiectasis and CF is beyond the scope of this review. Nevertheless, the treatment of each can be considered in terms of correcting the underlying cause where possible, which in CF now includes the possibility to correct or modulate the underlying defect. Thereafter management is directed towards treatment of bronchial infection, including oral and inhaled antibiotics for stable disease and prompt treatment of exacerbations, anti-inflammatory therapy, and chest clearance including physiotherapy and mucoactive drugs. For further information there are several reviews and guidelines on these topics [1, 2, 79].
Future directions
Bronchiectasis and CF are rapidly developing fields. The epidemiology of bronchiectasis is being increasingly understood with a large contribution from registry initiatives worldwide. The European Bronchiectasis Registry (EMBARC) is a European Respiratory Society initiative to stimulate clinical research in this neglected field and has operated a registry since early 2015 [102]. At the time of writing more than 3000 patients have been enrolled from more than 20 countries. Importantly, this initiative is also working with and supporting epidemiological research worldwide. This includes EMBARC India, the Indian bronchiectasis registry, which will generate the first population-based data on the characteristics and treatment of bronchiectasis in South Asia, and the Australian Lung Foundation registry, which will provide important data in both adults and children from Australasia. In the USA, the US NTM and bronchiectasis registry has enrolled more than 2000 patients at the time of writing [87]. Such international efforts are building on the success and contribution of US and European CF registries, which have been exceptionally important in understanding the natural history of CF and also in supporting randomised clinical trials of new therapies [103]. Ultimately the goal of all these epidemiological efforts must be to produce better standards of care and better treatments for our patients. An EMBARC task force recently reported on 22 research priorities for bronchiectasis agreed between expert physicians and patients aimed at driving such improvements. These included better epidemiology, improved provision of care and priority randomised clinical trials [104].
Conclusion
Bronchiectasis is a radiological abnormality that is associated with a wide range of clinical disorders including infectious, inflammatory and congenital disorders, including CF. Each individual disorder may be rare, but together the clinical problem is common and growing. All respiratory physicians should be aware of the clinical presentation and potential underlying causes of bronchiectasis in order to ensure appropriate early diagnosis and optimal treatment.
Self-evaluation questions
Regarding the aetiological diagnosis of bronchiectasis, which of the following is true?
a) Bronchiectasis affecting a single lobe is nearly always post-infectious
b) The only mandatory investigation for all bronchiectasis patients is serum immunoglobulin
c) CF should be excluded in all children and adults presenting with bronchiectasis
d) It is inappropriate to test for AATD in patients with bronchiectasis, regardless of the clinical presentation
e) NTM infection is strongly associated with bronchiectasis and it is important to rule out NTM in patients with bronchiectasis
Considering the impact of bronchiectasis, which of the following is false?
a) Exacerbations of bronchiectasis not due to CF account for a large proportion of the economic impact on the healthcare systems
b) As a result of the complex and multisystemic involvement of CF, treatment and follow-up care in specialist centres with multidisciplinary care teams is recommended.
c) The impact of bronchiectasis not due to CF is highly variable, with mild disease being characterised by infrequent exacerbations and the presence of P. aeruginosa colonisation
d) CF is no longer just the childhood disease it was years ago; today, the median survival is ∼40 years old
e) Airflow obstruction is associated with increased mortality in patients with non-CF bronchiectasis
A 26-year-old female presents for evaluation of a cough productive of thick mucus plugs, wheezing and difficult-to-control asthma. High-resolution CT demonstrates central bronchiectasis and bronchial wall thickening with mucus plugging to the right upper lobe. What is the test most likely to reveal the diagnosis?
a) Mannitol challenge test
b) Sweat chloride test and CF genetics
c) Serum total IgE and IgE to Aspergillus
d) Serum rheumatoid factor
e) Flexible bronchoscopy
A 37-year-old male presents with bilateral upper lobe bronchiectasis. He complains of persisting cough and large sputum volumes. He developed symptoms in his early 20’s and was unsuccessfully treated for asthma with inhaled corticosteroids. He is married with no children and works in a factory. He gets brief relief from antibiotics but symptoms rapidly recur. The most recent sputum culture grew S. aureus. He denies any other medical history. What is the most likely underlying diagnosis?
a) NTM infection
b) HIV infection
c) Asthma associated bronchiectasis
d) CF
e) Common variable immunodeficiency
Which of the following statements regarding geographical associations is not true?
a) NTM infection is more common as a cause of bronchiectasis in the USA than in Western Europe
b) The most common cause of bronchiectasis in India is post-infectious bronchiectasis
c) CF is a disease primarily affecting persons of European descent
d) The incidence of bronchiectasis is high in indigenous Australians
e) Bronchiectasis is underdiagnosed in East and South-east Asia compared with Europe and North America
Suggested answers
e
c
c
d
b
Footnotes
Support statement This work was supported by EMBARC, a European Respiratory Society Clinical Research Collaboration and EMBARC India, an initiative of EMBARC and the Respiratory Research Network of India (RRNI).
Conflict of interest Disclosures can be found alongside this article at breathe.ersjournals.com
- ©ERS 2016
Breathe articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References











Jump To
- Article
- Abstract
- Abstract
- What is bronchiectasis?
- What causes bronchiectasis?
- What are the links with “traditional” airway diseases (COPD and asthma)?
- What is the clinical presentation of bronchiectasis?
- Clinical impact of bronchiectasis
- Clinical impact of CF
- Bronchiectasis and CF in Europe
- Bronchiectasis and CF in North America
- Bronchiectasis and CF in Asia
- Management
- Future directions
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics