





Abstract
It is important to consider alternative diagnoses when a common respiratory condition presents with atypical features. In this patient, subtle systemic signs and an unusual clinical course hinted at an unexpected aetiology. http://bit.ly/2webytn
Background and initial presentation
A 32-year-old lady went into pre-term labour at 35 weeks gestation. She was admitted to hospital and received antenatal steroids. A female baby was born at 35+3 weeks, weighing 1.96 kg (9th centile on female UK–World Health Organization growth chart). She was delivered by emergency caesarean section due to fetal distress but required no resuscitation other than facial oxygen in the delivery room.
Antenatally, the 20-week ultrasound scan had revealed mild lateral ventriculomegaly. Fetal magnetic resonance imaging at 30+4 weeks gestation showed a mildly enlarged cisterna magna but normal cerebellar appearance. The radiologist noted nodularity of the ependymal lining of the lateral ventricles.
On admission to the neonatal unit, the baby had an oxygen requirement with mild respiratory distress but was otherwise well. On day 2 of life, she developed worsening respiratory distress and was commenced on continuous positive airway pressure (CPAP).
Task 1
What investigations would aid you in identifying the most likely diagnosis at this stage?
Answer 1
The most common causes for early respiratory distress in a neonate are transient tachypnoea of the newborn (TTN), respiratory distress syndrome (RDS), pneumothorax, sepsis and cardiac malformations. There are, of course, several other disorders to consider. Persistence of respiratory distress into day 2 makes TTN less likely. The relatively mature gestation and treatment with antenatal steroids reduces the risk of RDS but does not rule it out as a diagnosis. A stepwise approach to identify the most likely causes in the first instance is important.
Additionally, neonatal respiratory distress is one of the clinical indicators of early-onset sepsis[1]. In this case, it has presented in combination with spontaneous pre-term labour, which is an independent risk factor for early-onset sepsis. As a result, treatment for sepsis is indicated until negative culture results are returned.
A reasonable approach is as follows.
Septic screen: including full blood count, C-reactive protein (CRP) and blood cultures. Antibiotics should be commenced following collection of blood culture.
Chest radiography with nasogastric tube insitu: this may show signs of TTN, RDS, focal infective changes, pneumothorax or congenital malformations. Features of congenital lung or airway malformations may include cystic or multicystic areas, although in the neonate, lesions may be fluid filled, demonstrate air–fluid levels or appear solid. Large lesions may cause a mass effect. In some types of congenital heart diseases, chest radioraphy may show cardiomegaly and/or pulmonary plethora.
Full cardiac monitoring including pre- and post-ductal saturations with continuous ECG: a significant difference in pre- and post-ductal saturations may hint at a congenital cardiac defect (such as a coarctation of the aorta) or pulmonary hypertension.
Echocardiography: a formal echocardiogram is required to assess the function and structure of the heart in a baby without a clear respiratory cause for an ongoing oxygen requirement.
The baby was screened for sepsis, which revealed a low CRP and no growth on blood cultures. She received a short course of first-line empirical antibiotics. There were no other markers of sepsis, such as thrombocytopenia or coagulopathy. Her initial chest radiograph on day 1 showed slightly increased bronchovascular markings but was otherwise unremarkable (figure 1) and echocardiography showed only a small patent ductus arteriosus (PDA)with no structural abnormality or evidence of pulmonary hypertension.

Chest radiograph on day 1.
She required 24 h of CPAP, 5 days of humidified high-flow oxygen and then ongoing low-flow oxygen. Her repeat chest radiograph at 14 days' age revealed a ground-glass appearance of lung fields but this was on an expiratory film (figure 2). She never required invasive ventilation but remained in hospital for a total of 32 days, mainly due to a small but persisting oxygen requirement. At discharge, she was breastfeeding well but was still on 0.1 L·min−1 nasal cannula oxygen.

Chest radiograph on day 14.
Due to antenatally identified nodularity of the ependymal lining of the lateral cerebral ventricles, magnetic resonance imaging (MRI) of the brain was carried out prior to discharge.
Task 2
Figure 3 is an axial T2 section from the MRI brain scan carried out at 4 weeks of age. What abnormality is shown in this image?
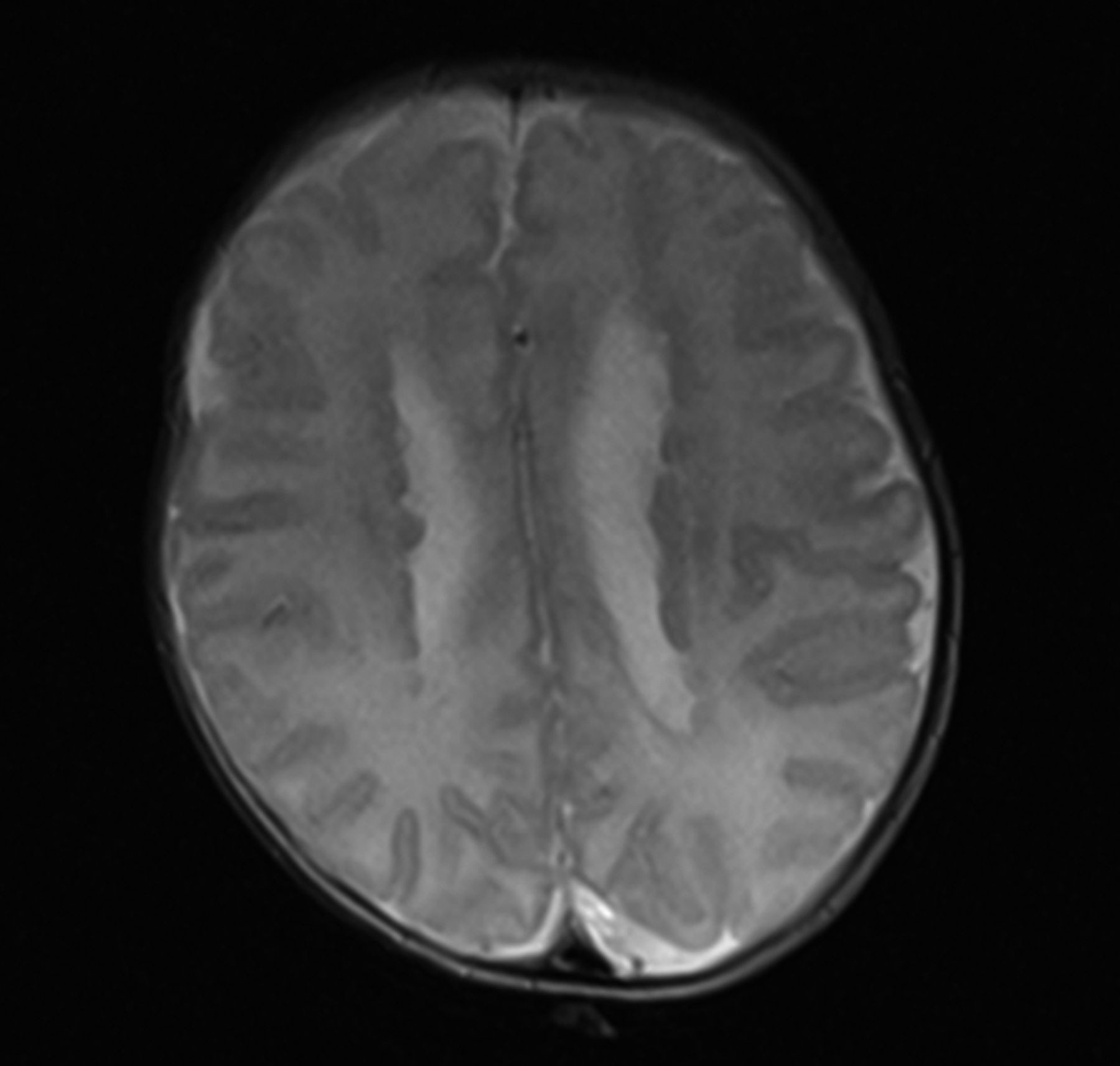
Axial T2 section from MRI brain scan at 4 weeks.
Answer 2
As suspected antenatally, nodules can be seen lining the lateral ventricles. This MRI demonstrates the typical appearance of periventricular nodular heterotopia (PNH). This occurs when neurons fail to migrate correctly between the sixth and 24th weeks of in utero development. In normal development, the neurons would migrate peripherally to the cerebral cortex. In PNH, some neurons remain as clumps in a more central position. PNH may be asymptomatic but is often associated with seizures in early to late childhood and is sometimes associated with learning difficulties [2].
Outpatient follow-up: neonatal clinic, 7 weeks
The patient was brought to the neonatal follow up clinic at 7 weeks of age. Although on minimal oxygen, there were some unanswered questions in her story. For a 35-week gestation baby with a relatively unremarkable neonatal course, a continued oxygen requirement was unusual.
The outcomes of isolated PNH can be unpredictable. Seizures and intellectual disability are the chief concerns. In this case, was the PNH isolated? Or was it related to the atypical lung disease?
Task 3
What additional investigations or referrals might be appropriate at this stage?
Answer 3
There is now evidence of a persisting respiratory problem with no clear identifiable cause. In addition, this baby has a cerebral abnormality that may or may not be linked to her respiratory concerns. It is possible that she will have additional learning needs or seizures. This family will require input from respiratory, genetics and community paediatric medical teams in order to manage her current symptoms, to investigate further for a unifying diagnosis, and to support the child and family living with a multisystem condition. Later on, she may require a neurology opinion although this is not clinically indicated at this point in time. It may also be reasonable to involve outreach nursing teams at this stage depending on the local provision.
At discharge, the baby was being cared for in a district general hospital. As a result, routine follow-up had not been arranged with the dedicated respiratory team for children with chronic lung disease of prematurity (CLDP). This service works closely with the neonatal team to manage CLDP as well as to explore more atypical features. Following this clinic review, the patient was referred to the respiratory team, as well as to community paediatrics and clinical genetics.
Outpatient follow-up: respiratory clinic, 3 months
The patient was seen in the respiratory clinic at 3 months of age. At this stage, she was on continuous nasal cannula oxygen and had mild respiratory distress when feeding. There were reported “choking” episodes during feeds, and she had a persistent cough that sounded wet and rattly, and was most obvious at night. The reason for her continued oxygen requirement was unclear.
She lived in a house free of mould and damp, and her parents and grandparents were nonsmokers. There was no relevant family history identified. Examination revealed suprasternal and subcostal recession as well as low muscle tone globally.
Task 4
Which of the following conditions would you consider in your differential diagnosis following this assessment?
Childhood interstitial lung disease (chILD)
Chronic aspiration
CLDP
Cystic pulmonary airway malformation (CPAM)
Pneumonia
Primary ciliary dyskinesia (PCD)
Spinal muscular atrophy (SMA)
Vascular ring
Answer 4
It is relatively easy to rule out pneumonia. The symptoms of pneumonia are highly unlikely to have such a prolonged duration and infection markers were not identified on initial screening. Regarding CPAM, there had been no abnormalities identified antenatally and post-natal chest radiographic changes were not consistent with this. A CPAM may result in a persistent oxygen requirement and progressive symptoms if the cystic lesion grows or compresses healthy lung tissue. A repeat chest radiograph could have provided a clue. However, an MRI or computed tomography (CT) scan of the chest is required to definitively exclude this as a diagnosis.
It is worth considering a neuromuscular disorder such as SMA, especially in the context of a child with low muscle tone and increased work of breathing. The timing and severity of the symptoms in this child are not typical of SMA. In SMA type 1, symptoms have an early onset and are severe, with very few active movements and a high likelihood of requiring mechanical ventilation. In the less severe SMA type 2, symptoms are milder but typically develop between 6 months and 1 year of age.
A vascular ring arising from a malpositioned blood vessel may compress or encircle the trachea, oesophagus or both. Symptoms can include increased work of breathing, biphasic stridor, cough and feeding difficulties. Although the patient displayed some of these symptoms, the oxygen requirement was unusual and the tell-tale stridor was lacking. CT or MRI would be required to image the thoracic anatomy, preceded by a barium swallow.
PCD should be considered as one of the differentials. Term respiratory distress and neonatal rhinorrhoea are the respiratory hallmarks of PCD. Cough and ear infections may appear later. The patient did not have rhinorrhoea although she did later develop a cough. She did not have dextrocardia/situs inversus either (although this is present in <50% of patients who have PCD). On balance, a diagnosis of PCD is unlikely.
CLDP, chronic aspiration (in the context of persistent cough and choking episodes) and chILD remain the top differentials.
Task 5
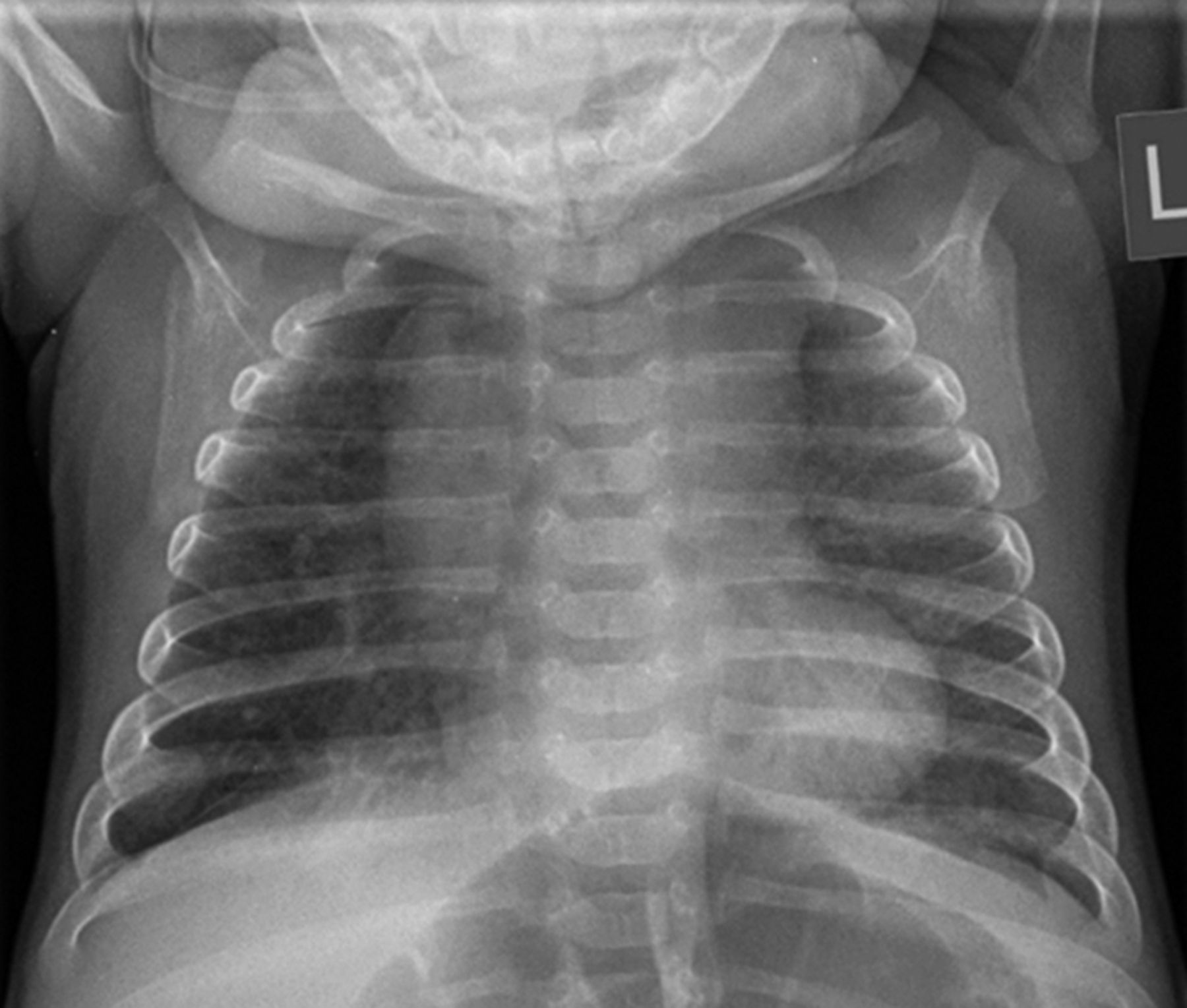
Does this chest radiograph taken at 3 months of age (figure 4) change your differential diagnoses?

Chest radiograph at 3 months.
Answer 5
This radiograph shows bilateral lung hyperinflation with increased interstitial markings throughout. This is usually consistent with a diagnosis of CLDP. There are no focal changes to suggest pneumonia or recent aspiration. There are no cystic areas and the cardiothymic silhouette is normal. In a neuromuscular disorder, one might expect to see a bell-shaped chest as a result of atrophy of the intercostal muscles and use of abdominal muscles in breathing. The chest cavity in this film is of normal shape other than being hyperinflated. This film suggests that CLDP or chILD are the most likely diagnoses.
Given her gestation and neonatal course, it was unlikely that this baby should have CLDP. As well as being atypical in gestation, the patient did not tolerate the standard CLDP oxygen weaning plan. The plan followed in our centre is that the first oximetry is undertaken 24–48 h after discharge home at the same flow rate as on the day of discharge. If the baby is clinically stable and growing, oximetry is repeated with reduction of oxygen by 0.1 L·min−1 every 3–5 weeks until 0.1 L·min−1 is reached, if the following target saturations are achieved:
Mean peripheral oxygen saturation (SpO2) ≥93%, preferably 95%, especially if pulmonary hypertension present
<5% of total study time at SpO2 <90%
Once the child is on 0.1 L·min−1 and has achieved a weight close to 5 kg, a nurse specialist performs a 2-h daytime study (including a period of feeding) in air. As long as SpO2 ≥93% when awake and feeding, a night-time air study is undertaken that night. A second air study is scheduled 10–12 weeks after the initial study. Oxygen equipment is removed after the infant has coped with at least one viral upper respiratory tract infection or at the end of winter.
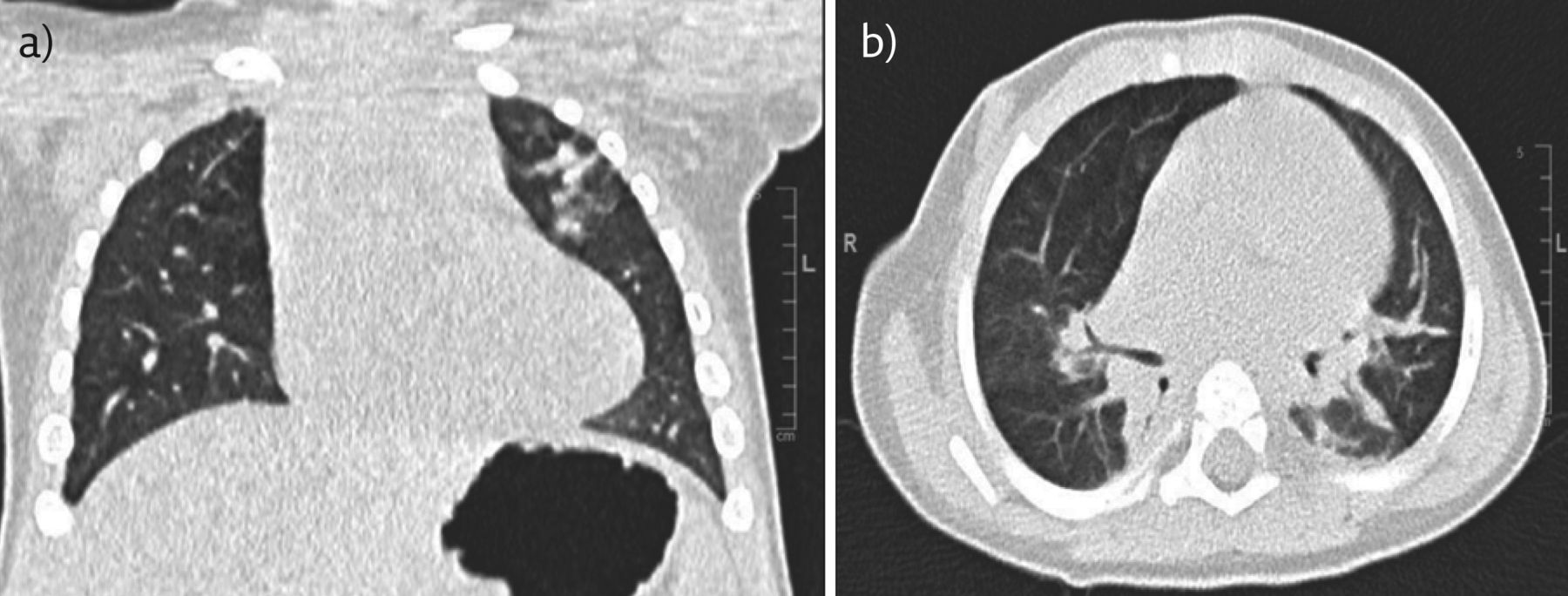
Her course was further complicated by a number of hospital admissions in the first 6 months of life, due to viral infections with respiratory syncytial virus, rhinovirus, adenovirus and enterovirus. There was no evidence, however, of bacterial infection on bronchoalveolar lavage. Bronchoscopy demonstrated structurally normal airways with no evidence of large airway malacia. Findings were corroborated by high-resolution CT (figure 5) performed at the age of 11 months, which showed normal lung parenchyma apart from general anaesthesia-related dependent changes. This would make the diagnosis of chILD less likely. There were no abnormalities identified on immune function testing.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a) Coronal and (b) axial sections from CT of the chest at 11 months.
In view of the persistent cough, respiratory distress and rattly breathing, an empirical sequential treatment trial was commenced. This consisted of inhaled beclomethasone, nebulised hypertonic saline, prophylactic azithromycin and regular airway clearance with physiotherapy. Over time, there was noticeable symptomatic improvement and oxygen requirement began to reduce. Neurodevelopmental assessment confirmed ongoing mild central hypotonia and motor delay but no other concerning features.
Outpatient follow up: genetics clinic, 6 months
PNH is a heterogeneous disorder, and can be observed in patients with chromosomal copy number variants and sequence variants in a number of genes. Despite increasing recognition of the genetic basis of this neuronal migration disorder, a specific genetic cause is not identified in a substantial proportion of patients. The patient's parents were unrelated and there was no family history of PNH or similar respiratory problems.
The patient had started smiling and parents were satisfied with her social development. She had not yet started rolling and mild central hypotonia was a persisting finding. She had greater than expected head lag on being pulled to sit. Previous chromosomal microarray analysis had retuned a normal result, excluding any pathogenic chromosomal deletions or duplications associated with PNH.
The association of loss-of-function variants in FLNA with genetically determined PNH in females is well-documented. FLNA is the gene which codes filamin A, an important cytoskeletal protein.
Task 6
Why are most patients with FLNA-related PNH female?
Answer 6
FLNA-related PNH is inherited in an X-linked dominant manner. Most affected patients are female as loss-of-function variants in FLNA are perinatally lethal in males.
Most females with FLNA-related PNH have epileptic seizures but intellect is usually normal. Extra-central nervous system manifestations, especially connective tissue abnormalities, including aortic dilatation, occur in a proportion of affected patients. A very rare association of unexplained chronic lung disease in patients with FLNA-related PNH had recently been reported in medical literature and, therefore, this diagnosis was considered [3].
Targeted analysis of the 47 FLNA coding exons (exons 2–48) by bidirectional fluorescent sequencing was requested. A heterozygous nonsense FLNA variant c.676C>T p.(Arg226*) was identified, compatible with FLNA-related PNH. Parental testing for this variant was negative, suggesting de novo origin of the variant in the patient.
FLNA gene encodes the protein filamin A, which regulates reorganisation of the actin cytoskeleton. This process is critical to the modulation of cell shape and migration. Loss-of-function variants in FLNA are known to affect migration of cerebral cortical neurons but the mechanism causing lung disease remains speculative. An FLNA knockout mouse model has been developed and a reduced lifespan of 3–4 months was noted in ∼20% of female heterozygous mice. Interestingly, lung oedema and emphysema were documented on autopsy in these mice [4]. Filamins may serve a mechanosensing role[5], and Burrage etal.[6] have plausibly suggested that disturbed mechanosensitivity could alter lung viscoelasticity and disturb alveolar modelling, while acknowledging that the exact mechanism of FLNA-related lung disease remains to be established.
Latest follow-up
Over time, there was clinical improvement and the patient was weaned off supplemental oxygen at the age of 1 year and 2 months. She remains off oxygen and is now 3.5 years old. She is on omeprazole for gastro-oesophageal reflux and prophylactic azithromycin. She has not had any seizures and is making appropriate neurodevelopmental progress. She has divarication of the recti, an umbilical hernia and hypermobile joints. There is no evidence of aortic root dilatation on echocardiographic screening, although she has minimal aortic regurgitation and a persistent smallPDA.
Discussion
The initial report postulating a causal relationship between loss-of-function FLNA variants and early-onset lung disease was published in 2011 [3]. There are now at least 23 reported cases of FLNA variants associated with interstitial lung disease in infancy (20 females and three males) [3, 6–18]. Table 1 lists the clinical features, investigation findings and outcomes of the reported patients. In all instances where MRI of the brain was performed, PNH was present in addition to lung disease. The skewed sex ratio reflects the X-linked dominant nature of FLNA loss of function.
Clinical features, genetic findings, imaging and pathology investigations, and outcome data on patients with early-onset (<12 months) FLNA-related lung disease
Despite the increasing reports, there are several unresolved questions about this unusual new addition to the phenotypic spectrum of pathogenic FLNA loss-of-function variants. While only loss-of-function (nonsense, frameshift and splicing) variants cause PNH with or without lung disease, there are no particular genotypes within the loss-of-function group predicting respiratory involvement. The severity of lung disease is also highly variable; ranging from our patient at the milder end of the spectrum to a series of six patients who required lung transplantation [6]. Reported histopathological features are diverse, including emphysema, pulmonary hypertension, atelectasis, cysts and varying degrees of alveolar simplification.
The true incidence of lung disease in patients with FLNA variants is unknown and it is likely that the 24 patients (including ours) significantly underascertain this. Over half of the published cases have been reported in the last 3 years, likely reflecting increasing recognition of the FLNA pulmonary phenotype. It is possible that children who succumb to severe respiratory illness in infancy are missed as they do not survive long enough to manifest seizures (prompting MRI brain) and at the other end of the spectrum, children with milder respiratory involvement, even with concomitant PNH, may not be diagnosed. In 2006, before the recognition of lung disease in this disorder, three pre-term siblings (twin boys and a female sibling) were reported by Gérard-Blanluet et al. [19] with PNH and “bronchodysplasia”.
The respiratory implications in adults are even less well understood. Clapham et al. [8] briefly mentioned a mildly affected mother with apical bullae and a maternal grandmother with lobar emphysema (family 3) where the female proband had died from respiratory failure at 7 months, and an affected male (case 29) in his late 30s considered for lung transplantation due to obstructive lung disease was reported as part of a large series by Lange et al. [20]. Much remains to be learned, therefore, about the respiratory outcome in older individuals, i.e. those who do not have respiratory problems in infancy. The FLNA variant in our patient was de novo but maternal inheritance has been described by others (table 1). There is little or no information on any history of respiratory problems in the mothers during childhood or later.
As more patients with FLNA variants and interstitial lung disease are reported, and the respiratory status of affected mothers and other affected female relatives is studied in greater detail, the natural history and underlying factors for this unusual cause of lung disease might be better understood.
Clinical implications
Our report adds to the growing number of children with FLNA-related lung disease. Given the relatively unremarkable CT findings in contrast to the more severe, progressive findings in previously published cases, it also adds to the phenotypic heterogeneity in FLNA-related lung disease.
MRI of the brain and/or FLNA analysis should be considered in children with unexplained chronic lung disease. While this will not be appropriate for all children, it is likely to be helpful where history, symptoms and/or radiological appearances are atypical, or there are unexpected associated features (e.g. in connective tissues). An atypical presentation and a course not consistent with CLDP in our patient prompted further assessments for alternative diagnoses. Clues in the history included antenatal scan findings and post-natal MRI of the brain. The subtle post-natal connective tissue findings (low muscle tone and hypermobile joints) were also relevant to the underlying diagnosis.
Footnotes
Conflict of interest: R. Walsh has nothing to disclose.
Conflict of interest: D. Batra was given a bursary to attend the Joint European Neonatal Society Conference in Venice by the British Association of Perinatal Medicine, which was funded by the Chiesi Pharmaceutical Company. This included travel and the conference fee.
Conflict of interest: A. Dixit has nothing to disclose.
Conflict of interest: J.M. Bhatt has nothing to disclose.
- Copyright ©ERS 2020
Breathe articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.










