





Abstract
Newborn bloodspot screening (NBS) for cystic fibrosis (CF) is an effective strategy for the early recognition of infants with a CF diagnosis. Some infants with a positive NBS result for CF have an inconclusive diagnosis and evidence suggests the number of these infants is increasing, as more extensive gene analysis is integrated into screening protocols. There is an internationally agreed, but complex, designation for infants with an unclear diagnosis after a positive screening result: cystic fibrosis transmembrane conductance regulator (CFTR)-related metabolic syndrome/cystic fibrosis screen positive, inconclusive diagnosis (CRMS/CFSPID). Infants with a CRMS/CFSPID designation have no clinical evidence of disease and do not meet the criteria for a CF diagnosis, but the NBS result indicates some risk of developing CF or a CFTR-related disorder. In this review, we describe the accurate designation of these and reflect on emerging management pathways, with particular attention given to clear and consistent communication.
Educational aims
To clarify the definition of the global harmonised designation: cystic fibrosis transmembrane conductance regulator-related metabolic syndrome (CRMS)/cystic fibrosis screen positive, inconclusive diagnosis (CFSPID).
To understand what impact a CRMS/CFSPID result has for the patient and their family.
Abstract
There is an internationally agreed, but complex, designation for infants with an unclear diagnosis after a positive screening result: CRMS/CFSPID. The majority of CRMS/CFSPID infants remain well and do not convert to a diagnosis of CF. https://bit.ly/3mtpJTu
Introduction
Newborn bloodspot screening (NBS) for cystic fibrosis (CF) has had a significant impact on the care of children with CF, improving nutrition and cognitive outcomes, reducing morbidity and maximising survival of these patients all over the world [1]. Since its development in the 1970s, there has been a steady global expansion of NBS for CF, especially in regions with a high incidence of CF. A wide variety of approaches for CF NBS have been used, but all result in occasional inconclusive diagnoses after a positive NBS result [2]. In most cases, a positive NBS result will translate into either recognition of carrier status or a straightforward diagnosis of CF; however, inconclusive diagnosis is becoming increasingly recognised leading to a challenging situation for both families and healthcare professionals [3]. Although most infants will remain healthy and asymptomatic, a small proportion may convert to a diagnosis of CF. As clinical experience with these cases grows, guidance with regards to the evaluation and management of these infants continues to evolve. Confusion still exists amongst clinicians with respect to the designation of these infants [4] and added to the uncertainty and distress for families, this clinical scenario is a challenging time for all those involved [5, 6]. It is therefore important to highlight the appropriate designation used for these infants, clarify the definition of the terminology used and consider the impact of this result on the journey of the child and their family.
The origin of the designation CFTR-related metabolic syndrome/CF screen positive, inconclusive diagnosis
There has been international variance with respect to the designation of infants with an inconclusive diagnosis after NBS for CF [3]. Given that infants in this situation are, by definition, healthy, it may be reasonable to consider whether a designation is necessary. The concern is that labelling healthy infants may expose them to an increased risk of unnecessary medicalisation and increased anxiety for the parents and families [7]. However, consensus groups in the USA and Europe decided that providing a designation would improve the patient pathway and facilitate the collection of data on these infants [8].
In 2009, a panel of experts completed a Delphi consensus exercise to produce the European consensus guidelines that provided an algorithm for the evaluation and early management of infants with an inconclusive or equivocal diagnosis following screening [9]. This guideline did not propose a designation, but instead focused on the need for clear communication with the family and what this situation meant for their child [2]. In the same year, a consensus group in the USA used a modified Delphi method to determine the designation cystic fibrosis transmembrane conductance regulator (CFTR)-related metabolic syndrome (CRMS) [8]. Although not a metabolic disease, the term was suggested as it was consistent with previous positioning of a CF diagnosis in the metabolic section of the International Statistical Classification of Disease code (ICD) database [7]. This provided a clear name for families that avoided using the words “cystic fibrosis” [1] and in the USA access to medical care is facilitated by a clear ICD code.
The CRMS designation was not widely accepted in Europe, as it was felt that parents would find the term confusing and use of “syndrome” suggested an underlying disease process [7]. Furthermore, using “metabolic” which denotes the elevated immunoreactive trypsinogen (IRT) is also not appropriate in this situation [3]. A further Delphi consensus process was undertaken in Europe to better guide the early management of infants with an unclear diagnosis following a positive NBS result. During that exercise there was agreement that by giving a name to this situation, a more consistent approach to management and data collection could be achieved. In a separate exercise, a wide range of terms, including CRMS, were considered. From this exercise the designation “CF screen positive, inconclusive diagnosis” (CFSPID) was adopted in Europe [2].
The European consensus emphasised that, although the abbreviation was more to provide a clear description of the situation, providing a “label” would be important to some healthcare systems in order to activate the right services and support for patients [2]. They also highlighted that providing a clear classification would enhance the communication between families and professionals while supporting future epidemiological analysis about the outcomes for this group of patients [10].
With respect to management guidance, CFSPID infants were divided into two groups: Group A, those with a normal sweat chloride (<30 mmol·L−1) and two CFTR gene variants (at least one of which has unclear phenotypic consequences); and Group B, those with an intermediate sweat chloride (30–59 mmol·L−1) and one or zero CFTR gene variants [2, 3]. In dividing infants like this, they recognised that the guidelines for early management and follow-up were slightly different for each group, reflecting a higher level of concern for those infants with an intermediate sweat chloride (Group B) [2].
The main difference between the CRMS and CFSPID designations related to infants with two CFTR gene variants and an intermediate sweat chloride. In the USA, these infants were designated as CRMS, whereas in the European guidance it was felt that these infants should move towards full CF care.
There was recognition that this inconsistent approach was not ideal and at the 28th North American CF Conference in Phoenix an international diagnosis consensus meeting was held [11]. From that meeting the harmonised designation of CRMS/CFSPID was proposed and this included infants with an intermediate sweat chloride value and two CFTR gene variants, if one or both variants were not characterised as CF causing (table 1).
Definitions of CRMS, CFSPID and the global harmonised designation CRMS/CFSPID
This designation now supersedes both CRMS and CFSPID, allowing collection of data on infants from all over the world to be combined, optimising comparisons between cohorts and increasing our understanding of how these patients evolve long term [5]. In fact, the planning committee reviewed data from Europe, Canada, Australia and the USA, providing additional guidance on further testing for CRMS/CFSPID patients and recommendations for the follow-up of these infants [7].
After the global harmonised designation, updated guidance on the management of CRMS/CFSPID patients was published [3]. Based on the increasing evidence available, this best practice guideline guides physicians to provide more consistent care pathways and achieve optimal outcomes.
Identifying CRMS/CFSPID infants
CRMS/CFSPID is identified following CF NBS when high levels of IRT are detected on the dried blood sample obtained in the first week of life. There are a number of second tier approaches that can then be undertaken to improve the performance of the protocol, most commonly testing for CFTR gene variants [12]. Over 2000 CFTR gene variants have been identified, but not all have been characterised as CF causing [13]. The Clinical and Functional Translation of CFTR (CFTR2) project is an international initiative to improve the characterisation of CFTR gene variants through cross-sectional epidemiological data from a large number of CF patients combined with in vitro data (available via a regularly updated website: https://cftr2.org). The project has defined the functional and phenotypic consequences of over 400 CFTR variants, helping to reduce the number of infants with an indeterminate diagnosis [1]. The harmonised definition CRMS/CFSPID acknowledges the increased capacity of the CFTR2 project over the past decade and incorporates the knowledge that CFTR variants are classified into four categories (table 2) [3, 5]:
“CF causing”
“non-CF causing”
“variant of varying clinical consequence (VVCC)”
“variant of unknown significance (VUS)”
CFTR2 classification of CFTR variants
The main difference between the harmonised definition CRMS/CFSPID and that of the separate terms CRMS and CFSPID, is the inclusion of the more specific term “CF causing variant” for infants with an intermediate sweat chloride [4]. As described above, for the initial European definition of CFSPID, infants with two CFTR gene variants (including those which would now be identified as VVCC or VUS) and an intermediate sweat chloride would be diagnosed as CF, whereas now an infant designated CRMS/CFSPID may have two CFTR variants and an intermediate sweat chloride if one of the variants is a VVCC or VUS (figure 1) [3].

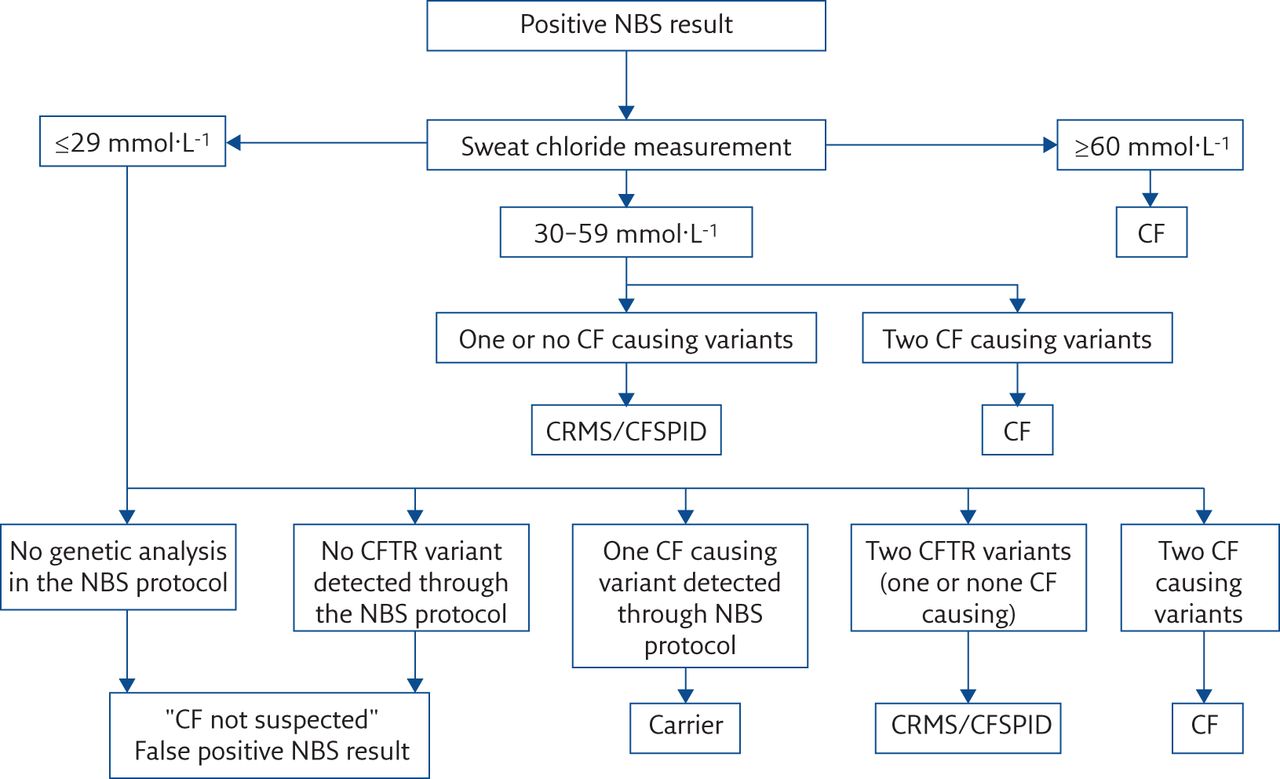{kind=link}
Algorithm for the designation of infants following a positive NBS. Reproduced and modified from [4] with permission.
The classification of CFTR gene variants provides some clarity for CF diagnosis, especially for variants that are associated with less severe phenotypes, but unfortunately the majority of CFTR variants are yet to be characterised [1]. For many variants this will never be possible as they do not occur in sufficient numbers and in vitro testing is not completely reliable. Consequently, there will always be infants with a positive CF NBS where genetic testing may still lead to an unclear diagnosis. As well as the absolute requirement for sweat testing as the gold standard bioassay for any infant with a positive CF NBS, other electrophysiological tests have been identified which may provide functional analysis of CFTR activity [3]. Based on in vivo evaluation of abnormal ion transport secondary to CFTR dysfunction in the nasal epithelial channels, testing of nasal potential difference (NPD) may allow infants with CRMS/CFSPID and CFTR dysfunction to be discriminated from those with normal CFTR function, thereby identifying those who are more likely to be diagnosed as CF in the future [10]. However, NPD is extremely challenging to undertake in preschool-age children and apart from its use in certain highly specialised CF centres [14], it is not currently used in clinical practice. Similarly, ex vivo intestinal current measurement of CFTR function (ICM) on rectal biopsies and intestinal organoid analysis are both specialised tests with limited availability. They may have a role as a CFTR biomarker [3, 5]; however, due to insufficient evidence of their validity they are not recommended in the routine assessment of CRMS/CFSPID infants [10].
What does a designation of CRMS/CFSPID mean for the healthcare system?
Although some diverse practice exists with regards to the management of CRMS/CFSPID infants, one consistent theme is the need to avoid over-medicalisation [4]. A small proportion of infants designated CRMS/CFSPID may eventually develop clinical features consistent with a diagnosis of CF [15]. Some children may convert to a CF diagnosis if their sweat chloride concentration increases into the CF diagnostic range. Therefore, regular review by clinicians with an interest in CF should be undertaken so that opportunities to provide the appropriate treatment are not missed.
There has been progress over the past 10 years with regards to the evaluation and early management of these patients based on emerging evidence and consensus of the Neonatal Screening Working Group core committee [3]. There is some debate on the total length of follow-up necessary and what the overall management of these patients should be as they go through childhood. The latest European guidance reflects a balanced approach between avoidance of over-medicalisation and the need for careful monitoring (table 3) [5].
Evaluation and follow-up investigations of CRMS/CFSPID infants
Initial assessment
At the initial assessment, investigations should include a repeat sweat test, extended CFTR gene analysis (check for updates in https://cftr2.org), stool for faecal elastase analysis, and clinical evaluation of the respiratory and gastrointestinal systems (history of cough/recurrent infections, respiratory and abdominal examination, weight, height, BMI).
CRMS/CFSPID infants can be managed in either a CF centre or a non-CF clinic environment by a CF specialist, depending on local resources [2, 3]. It is important that these consultations are organised in a manner that reduces any risk of cross infection with CF pathogens [7, 8]. A dedicated CRMS/CFSPID clinic may the ideal practice, but this may not be possible given the small numbers of CRMS/CFSPID infants at each CF centre [10].
For discussion with parents, most health professionals would use the term CFSPID, as the full name is complex.
Monitoring in the first 2 years
Frequency of follow-up clinics in the first 2 years is dependent on the clinical progress of the infant, parental anxieties, and the framework of the individual tertiary centre where the patients will be seen [2, 8]. This may include 6-monthly visits initially being reduced to annual reviews if the child and family are doing well [10]. Prospective studies have shown that sweat testing should be repeated at 6 months and 2 years of age [15]. There is no evidence to support the use of routine respiratory culture or imaging for infants who remain healthy [16] and further investigations should only be considered if clinically indicated. Faecal elastase can fluctuate in the first year of life in infants with CF, so if there are concerning features (weight loss) repeating this test may be appropriate [3].
Monitoring in preschool-age children (3–5 years)
Preschool-age children should be seen at least annually for full clinical evaluation of the respiratory and gastrointestinal systems (including height and weight). As new information on CFTR gene variants is available, an annual review gives the opportunity to revisit variants using CFTR2. Further investigations at this stage should then be guided by the clinical progress and if there are concerns, a repeat sweat test should be organised. If the sweat chloride increases into the CF diagnostic range in an asymptomatic child, it must be repeated again to confirm a CF diagnosis [3]. As the infant grows, there is no evidence to support routine respiratory culture, imaging or measurements of lung function (including LCI) in those who are well.
Any new results or genetic information must be fully discussed with the family so communication remains consistent and their questions are addressed. Clinicians involved in the monitoring of these children should ensure that detailed reports/letters are sent to the child's general practitioner (GP), which should also outline the symptoms that would need further assessment or discussion with the CF team (persistent/productive cough, problems with weight gain or bowel movements). It is important that children with a CRMS/CFSPID designation are included on an appropriate national database to monitor outcomes in this group and to better characterise the natural history of patients with a CRMS/CFSPID designation [5].
Assessment at 6 years
The most recent European guidance recommends that CRMS/CFSPID children should undergo a more detailed assessment when they are 6 years of age [3]. Investigations should include a repeat sweat test, measurements of lung function (LCI via multiple breath washout and if possible, incentive spirometry) and chest imaging (chest radiography or limited high-resolution computed tomography after discussion of the risks of radiation with the parents). If clinically indicated further extended CFTR gene analysis, faecal elastase and respiratory culture should also be considered at this evaluation and once again, any updates on CFTR2 should be checked.
Once the child has been clinically assessed and information from these investigations is available, parents should be invited to discuss options regarding future management and follow-up. Studies have shown that CRMS/CFSPID children at 6 years of age who remain healthy with a normal sweat chloride, normal growth and lung function/imaging are unlikely to convert to a CF diagnosis [16]. A small number of children may be diagnosed with CF following this more detailed assessment, but it is more likely that the information will provide further reassurance that the child is healthy and will likely remain healthy [3]. It is therefore reasonable to give the parents the option of being discharged from the clinic.
The parents and the GP should be informed that if any prolonged respiratory or abdominal symptoms occur these should be discussed with the CF team promptly and a route to do this should be provided. For some families, discharge from services may not be considered appropriate and they should be given the opportunity to continue regular reviews with the CF team or be seen in a respiratory clinic.
For well children who are discharged after the review at 6 years of age, it may be appropriate to organise a follow-up appointment. This will require some logistical planning but should be at an age when the consultation can be directed at the young person (see below).
Assessment in adolescence (14–16 years)
Assessment at this stage not only gives the opportunity to undertake a full review of the young person's clinical progress, but also enables direct engagement with the patient to discuss the long-term implications of the CRMS/CFSPID designation including CFTR-RD and what it means for them. Topics such as male infertility secondary to congenital bilateral absence of the vas deferens (CBAVD) and what further investigations may be required in early adulthood (semen analysis, ultrasound imaging of the testes for the vas deferens) can be discussed if the parents feel this is appropriate [3]. Advice on healthy living, in the context of a priori risk, can also be reinforced during this review, such as not smoking, maintaining a balanced diet and staying active. Signposting young people to reliable online resources, providing written information and a point of contact for the future will ensure that the young person has the appropriate support as they move forward in their life.
The risks of developing a CFTR-related disorder
A CFTR-RD was defined in 2011 as a clinical entity associated with CFTR dysfunction that does not fulfil the diagnostic criteria for CF [17]. Although, to some degree, this represented a diagnosis of exclusion, it did represent a paradigm shift and one that enabled a broader perspective of the impact of CFTR gene variants. In light of progress in both molecular genetics and CFTR biomarker work, the CFTR-RD definition is being revisited by a European CF Society working group, with the aim of better characterising this entity.
The conditions that are most well characterised as CFTR-RDs are CBAVD (a cause of male infertility) and (recurrent acute or chronic) pancreatitis [18]. A CFTR-RD relates to a clinical presentation and, as such, is quite distinct from the CRMS/CFSPID designation. It is clear that CFTR gene variants reported in those with CFTR-RD are regularly found in infants with CRMS/CFSPID [4]. The risk of a child designated CRMS/CFSPID developing CFTR-RD has not been fully quantified.
This is a difficult concept of risk that needs to be expressed to the families and eventually the person with a CRMS/CFSPID designation. There is some guidance in the recent European paper on what information might be shared with an individual [3]. With clear information, there is potential to facilitate appropriate care and avoid harm (for example, for a young man with CBAVD, knowledge of the risk may prevent a distressing journey and facilitate therapeutic options).
What does the designation of CRMS/CFSPID mean for the family?
With CRMS/CFSPID recognised as an outcome of CF NBS, healthcare professionals need to be prepared to counsel the families of these infants. Uncertainty, distress and anxiety begin for these families from the moment the NBS result is identified as positive, compounded further by the additional tests required (sweat test) in order to confirm or refute a definitive diagnosis [3]. The negative psychological impact for families of CRMS/CFSPID infants is increased further because the inconclusive result will be difficult for most parents to understand or be incongruent with their preconceived healthcare beliefs [6]. Although there is no internationally accepted approach to counselling families, it is essential that they are provided with clear and concise information on all steps of the process (table 4).
Communication with families of infants identified as CRMS/CFSPID
Parents should understand that infants with CRMS/CFSPID are well, and most will remain well. Information should also be provided about those infants that may eventually be diagnosed with CF, which can occur if their sweat chloride concentration increases into the CF diagnostic range, their CFTR variant is reclassified as disease causing or if significant symptoms develop [7]. Parents should be informed of the schedule for follow-up and a clear explanation provided about the plan for investigations and assessment, including a more detailed evaluation when their child is 6 years old.
Consistent information from various members of the multidisciplinary CF team is essential to maintain trust in the team and the process. It is important to know that for most families, the CRMS/CFSPID designation is not considered a good outcome and therefore it should not be presented as “good news” compared with a definitive CF diagnosis [3]. Anxiety triggered by a CRMS/CFSPID result has been shown to endure for parents, many of whom feel unsettled or continue to believe their child has CF [6]. Tensions therefore may arise between the views of the family and healthcare professionals in the understanding of the CRMS/CFSPID designation. It is important that these are recognised and resolved in order to minimise the negative psychological impact of the result.
As a key member of the multidisciplinary CF team, clinical psychologists offer essential support to families and eventually the patient themselves through their journey. Families of infants identified as CRMS/CFSPID should also be referred for genetic counselling as this offers another avenue of support. The counselling should include: documentation of family history, carrier status education, family planning discussion, and in some cases parental genotype testing may be necessary [12]. Evaluation of siblings should also be discussed with the family and, if appropriate, DNA analysis of other children in the family may be considered [3].
Conclusion
There is increasing information on the outlook for infants with a CRMS/CFSPID designation. A more consistent international approach to definition and designation has been a big step forward in the management of these infants, even if the harmonised term is rather unwieldy.
The approach to screening impacts on the frequency of CRMS/CFSPID recognition and the outlook for these infants. There is clear evidence that infants with an intermediate sweat chloride value are more likely to convert to a CF diagnosis through a rise in subsequent sweat values or clinical features. The longer-term outlook for these infants is much less clear, especially the risk of developing a CFTR-RD. There is good evidence to suggest that children with CRMS/CFSPID who are well at 6 years of age are extremely unlikely to convert to a CF diagnosis and this formed the basis of new European guidance that, following a detailed and reassuring evaluation at this age, families may be discharged from regular review, on the assurance that they are provided clear information and the opportunity for a follow-up visit at some later stage (possibly 14 years of age). As with all aspects of this situation, clear and consistent information for the family and the person with a CRMS/CFSPID designation is key.
Key points
There has been international variance with respect to the designation of infants with an inconclusive diagnosis after positive NBS.
The global harmonised designation CRMS/CFSPID now supersedes both CRMS (USA) and CFSPID (Europe).
Progress in the field has partly reflected improved classification of CFTR gene variants.
Most CRMS/CFSPID infants remain well and do not convert to a diagnosis of CF.
Some may convert to a CF diagnosis, most often because their sweat test result becomes positive.
The psychological impact associated with CRMS/CFSPID is challenging and highlights the need for consistent and clear communication between families, patients and healthcare professionals.
Self-evaluation questions
What is the term and abbreviation used as a designation for infants with a positive NBS result but an unclear diagnosis of CF?
What is the most important component of the management of infants with this designation?
What is the commonest reason for conversion from a CRMS/CFSPID designation to a CF diagnosis?
For infants with CRMS/CFSPID who do not convert to a CF diagnosis, what other condition are they at risk of developing when older?
Suggested answers
CFTR-related metabolic syndrome/CF screen positive, inconclusive diagnosis (CRMS/CFSPID).
Clear and consistent communication with the family.
Evolving sweat test result into the CF range (sweat chloride >59 mmol·L−1).
A CFTR-related disorder.
Footnotes
Conflict of interest: A. Sinha has nothing to disclose. K.W. Southern has nothing to disclose.
- Received May 28, 2021.
- Accepted August 11, 2021.
- Copyright ©ERS 2021
Breathe articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.











Jump To
- Article
- Abstract
- Abstract
- Introduction
- The origin of the designation CFTR-related metabolic syndrome/CF screen positive, inconclusive diagnosis
- Identifying CRMS/CFSPID infants
- What does a designation of CRMS/CFSPID mean for the healthcare system?
- The risks of developing a CFTR-related disorder
- What does the designation of CRMS/CFSPID mean for the family?
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics