
Abstract
Background and objectives: Armodafinil R-modafinil) is the R- and longer-lasting isomer of the racemic compound modafinil, a wakefulness-promoting medication. Armodafinil is eliminated approximately three times more slowly than the S-isomer of racemic modafinil. Published studies have demonstrated the efficacy of armodafinil for treating excessive sleepiness associated with obstructive sleep apnoea, shift work disorder and narcolepsy. The objectives of this study were to describe the pharmacokinetic profile, tolerability and safety of armodafinil in healthy subjects.
Methods: Pooled pharmacokinetic data from three separate randomized studies in 119 healthy subjects who received single or multiple (once daily for up to 14 days) oral doses of armodafinil ranging between 50 and 400 mg were analysed. The impact of food on the single-dose pharmacokinetic profile of armodafinil was also assessed in subjects following an overnight fast and after the consumption of a standard fatty meal.
Results: Armodafinil was readily absorbed and exhibited linear pharmacokinetics over the 50–400 mg dose range. Peak plasma concentrations were reached around 2 hours after administration in the fasted state. Food had no effect on the overall bioavailability of armodafinil; however, the peak concentration was delayed by approximately 2–4 hours. In the multiple-dose study, dose proportionality was confirmed by linear regression analyses of the log-transformed area under the plasma concentration versus time curve (AUC) and maximum plasma concentration (Cmax) values as a function of dose. After reaching the peak, plasma concentrations of armodafinil declined in a monophasic manner, with a mean elimination half-life of approximately 15 hours. Steady state appeared to be reached within 7 days. At steady state, the systemic exposure to armodafinil was 1.8 times that observed after single-dose administration. Armodafinil was generally well tolerated, the most frequent adverse events being headache, dizziness and nausea.
Conclusions: In the present analysis, armodafinil exhibited linear pharmacokinetics over the dose range of 50–400 mg. While food affected the rate but not the extent of absorption, peak plasma concentrations were reached in approximately 2 hours when the drug was taken on an empty stomach. With once-daily dosing, steady state appeared to be reached within 7 days. After reaching peak plasma levels, concentrations of armodafinil declined monophasically, with a mean elimination half-life of around 15 hours. Armodafinil was generally well tolerated.
Similar content being viewed by others
Background
Armodafinil (R-modafinil) is the longer-lived of the two isomers composing the racemic compound modafinil, a wakefulness-promoting medication. Armodafinil has been shown to improve wakefulness significantly in patients with excessive sleepiness associated with obstructive sleep apnoea, shift work disorder and narcolepsy.[1–4] The longer-lasting R-isomer was developed in an effort to improve upon the pharmacokinetic profile of modafinil, which has effects that are not always maintained throughout the day with once-daily administration, making it necessary either to increase the dose and/or to split the dose between morning and midday (undermining the convenience of once-daily administration) in order to obtain effective control of excessive sleepiness later in the period patients are awake.[5–7]
Previous studies have shown that the R- and S-isomers of modafinil have an approximately equipotent pharmacological activity,[8] but different pharmacokinetic profiles. The elimination half-life (t½ β) of R-modafinil (the levorotatory isomer) is approximately 15 hours in humans, whereas that of S-modafinil (the dextrorotatory isomer) is approximately 4 hours. This stereospecific difference in the clearance of its two isomers results in a biphasic decline in modafinil plasma concentrations from peak levels.[9,10] As a result of the slower (i.e. monophasic) clearance of the R-isomer, it was hypothesized that armodafinil might maintain effective plasma concentrations for longer periods than modafinil with once-daily oral administration. In a comparison of the pharmacokinetic and pharmacodynamic effects of armodafinil and modafinil in healthy subjects undergoing acute sleep deprivation, doses of armodafinil 200 mg achieved comparable peak plasma concentrations but higher concentrations at 6–14 hours after administration than modafinil 200 mg, as well as increased wakefulness and attention performance for longer periods post-dosing.[11]
The present analysis was undertaken to further characterize the pharmacokinetic profile of armodafinil and to examine its tolerability and safety in healthy volunteers. The pharmacokinetic profile described in this analysis is derived from data compiled from three randomized studies in healthy subjects who received single (with or without food) or multiple oral doses of armodafinil ranging from 50 mg to 400 mg.
Subjects and Methods
The three pharmacokinetic studies were randomized investigations conducted either in the UK (studies 1 and 2) or the US (study 3). All three were approved by the independent ethics committees/institutional review boards at the respective study sites, and were conducted in accordance with International Conference on Harmonisation (ICH) good clinical practice guidelines,[12] and applicable national and local laws. Written consent was obtained from each subject before any procedures or assessments were performed and after the aims, methods and potential hazards had been explained.
The demographic characteristics of the subjects in the three studies were similar, as were the entry criteria for enrolment and the methodologies used to measure the concentrations of armodafinil and metabolites in plasma and to determine the drug’s pharmacokinetic parameters. For these reasons, pooling of data from these studies was considered acceptable.
Study Design and Dosing
Studies 1 and 2 were randomized, double-blind, placebo-controlled, parallel-group investigations of the pharmacokinetics and tolerability of armodafinil in healthy men. In both studies, randomization was performed using a randomization code, while blinding was achieved by matching placebo to 50 mg capsules of the study drug. Each investigator was accountable for the study drug and placebo throughout the study.
Subjects enrolled in study 1 were randomized to receive a single dose of armodafinil 50, 100, 200, 300 or 400 mg or matching placebo in the morning after an overnight fast. Those who received the 100 mg dose returned to the clinic after 1 week to receive a second 100 mg dose after consuming a standard fatty meal.
Subjects enrolled in study 2 were randomized to receive armodafinil 50, 100, 200, 300 or 400 mg or matching placebo once daily for 14 consecutive days. Subjects received armodafinil each morning after an overnight fast either as inpatients (on days 1, 7 and 14) or outpatients. Blinded assessment of the safety data for the 200, 300 and 400 mg dose groups indicated that 400 mg appeared to be an intolerable dose and this dose was discontinued after 7 days of administration. Because 200 and 300 mg doses appeared to be well tolerated, an intermediate dose of 250 mg was added to further evaluate the tolerability profile of armodafinil. Subjects in this dose group followed the same experimental procedures as those used for the initial dose groups.
Study 3 was a randomized, open-label, crossover investigation to compare the bioavailability of the highest dosage strength of uncoated tablets planned for commercialization versus that of an equivalent dose of multiple 50 mg coated tablets used during early clinical testing. Healthy men or women received a single dose of armodafinil as 5 × 50 mg coated tablets or as 1 × 250 mg uncoated tablet. Subjects were randomized to receive either dose in the morning after an overnight fast. Following a 7-day washout period, the alternative dose was administered, again after an overnight fast.
In all three studies, subjects were not permitted to take any other prescription or over-the-counter medications (with the exception of paracetamol [acetaminophen]) or alcohol within 48 hours before each administration of the study medication or throughout each pharmacokinetic sampling period.
Subjects
Studies 1 and 2 were to be conducted in men aged 21–40 years who were in good health and had a body mass index (BMI) ≤30 kg/m2. Exclusion criteria were: (i) clinically significant, uncontrolled medical conditions; (ii) abnormal laboratory values, vital signs, ECG or physical examination findings; (iii) a history of smoking or alcohol or drug abuse, or excessive consumption of caffeine-containing beverages; (iv) positive test results for hepatitis B surface antigen or hepatitis C antibodies; or (v) a history of HIV-positive status.
Study 3 was to be conducted in men or women aged 18–45 years who were in good health and had a BMI ≤30 kg/m2. The exclusion criteria were similar to those for studies 1 and 2; women enrolled in the study were required to be surgically sterile, 2 years postmenopausal, or using a medically accepted method of birth control if they were of child-bearing potential. Steroidal contraceptives had to be used in conjunction with a barrier method.
Pharmacokinetic Assessments
In all three studies, plasma samples were collected before armodafinil administration on day 1 and at prespecified times after administration. In study 1, plasma samples were collected at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 13, 16, 24, 36, 48, 72 and 96 hours after drug administration. In study 2 (multiple-dose study), plasma samples were collected on days 1, 7 and 14 at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 13, 16 and 24 hours after drug administration, with additional samples at 48, 72 and 96 hours after the last dose on day 14. In addition, pre-dose samples for estimation of trough concentrations were collected on days 5, 6, 7, 12, 13 and 14. In study 3, plasma samples were collected at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 13, 16, 24, 36, 48, 60 and 72 hours after administration of the first dose, and at the same times following the alternative regimen administered after the 7-day washout period.
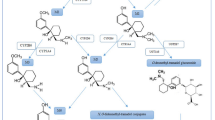
Plasma samples from studies 1 and 2 were analysed for concentrations of armodafinil and its two major circulating metabolites, R-modafinil acid and modafinil sulfone (figure 1), by the Department of Drug Safety and Disposition of Cephalon, Inc. (West Chester, PA, USA), using a validated high-performance liquid chromatography (HPLC) method with UV detection. The assay utilized liquid-liquid extraction of armodafinil and its two circulating metabolites from 0.20 mL of plasma to which had previously been added the internal standard, (phenylthio)acetic acid. After mixing and centrifugation, the samples were placed on dry ice until the aqueous layer had frozen. The organic layer was then poured into a clean glass centrifuge tube, and the solvent was evaporated under a stream of N2. The dried residue was reconstituted in 0.20 mL of acetonitrile: 0.02 mol/L KH2PO4 buffer, pH 2.5, 30: 70 (v/v), and 30 µL was injected onto a ® BDS phenyl column (2.1 × 150 mm; 5 µm particle size) maintained at 35°C. Chromatography was isocratic, with a mobile phase consisting of a 32: 68 ratio of methanol: 0.02 mol/L KH2PO4 buffer, pH 2.5 (v/v). Elution during the 24-minute run was monitored at 225 nm; armodafinil, R-modafinil acid, modafinil sulfone and the internal standard eluted at approximately 12.5, 14.5, 15.5 and 6.8 minutes, respectively. Chiral chromatography was not required because there is no interconversion of armodafinil or R-modafinil acid with its corresponding S-isomer. Within-run and between-run coefficients of variation over the assay range (0.20–50.0 µg/mL for each analyte) were ≤4.9% and ≤11.1%, respectively. Within-run accuracy and between-run accuracy were 87.9–107% and 92.9–101%, respectively.

Structural configuration of armodafinil and its two major circulating metabolites, R-modafinil acid and modafinil sulfone.
Plasma samples from study 3 were analysed for concentrations of armodafinil, R-modafinil acid and modafinil sulfone by PPD Development (Middleton, WI, USA) using a similar, validated HPLC-UV method. Sample processing was conducted the same way as outlined above, except that the internal standard was CEP-20253, a structural analogue of modafinil. Chromatography was performed on a Zorbax® Eclipse XDB-C18 column (2.1 × 30 mm; 3.5 µm particle size) with monitoring of analyte elution at 235 nm. Armodafinil, R-modafinil acid, modafinil sulfone and the internal standard eluted at approximately 3.6, 5.1, 6.5 and 10.5 minutes, respectively. Assay precision and accuracy over the assay range (0.20–50.0 µg/mL for each analyte) were consistent with those reported above.
Noncompartmental pharmacokinetic analysis was performed for all three studies. Standard pharmacokinetic parameters of armodafinil, R-modafinil acid and modafinil sulfone after single and multiple doses were determined, as applicable, using WinNonlin® (Enterprise Versions 4.0.1 or 4.1; Pharsight Corporation, Mountain View, CA, USA), including: (i) the maximum plasma concentration (Cmax); (ii) time to Cmax (tmax); (iii) minimum (trough) plasma concentration in a dosing interval (Cmin); (iv) area under the plasma concentration versus time curve (AUC) from time zero to infinity (AUC∞) for a single dose and AUC over one dosing interval (τ) for the multiple-dose regimen (AUCτ); (v) apparent total oral clearance (CL/F); (vi) t½ β; and (vii) apparent volume of distribution (Vz/F). In study 2, the steady state and observed accumulation ratios (Rss and Robs, respectively) were determined as the ratio of AUCτ on days 7 or 14 to AUC∞ on day 1 (Rss) or to AUC from time zero to 24 hours (AUC24) on day 1 (Robs).
Where possible, standard pharmacokinetic parameters of R-modafinil acid and modafinil sulfone were also determined in all three studies. Dose-normalization of pharmacokinetic parameters for armodafinil and its metabolites to a 50 mg dose was performed through a proportional adjustment of the values for each subject by multiplying by the appropriate dose-normalization factor (i.e. 50 mg/dose from which the value was derived) and then determining the mean value.
Tolerability/Safety Assessments
The tolerability of armodafinil was assessed in each study by an adverse event inquiry each time a blood sample was collected. Safety was evaluated by monitoring clinical laboratory test results, vital sign measurements, ECG and physical examination findings. The latter assessments were performed at baseline and at regular intervals after armodafinil administration (i.e. at each pharmacokinetic sample collection in the case of vital signs and ECG, and at the conclusion of the study in the case of clinical laboratory tests and physical examinations). Adverse events were classified according to the Coding Symbols for a Thesaurus of Adverse Reaction Terms (COSTART)[13] in studies 1 and 2, and version 7 of the Medical Dictionary for Regulatory Activities (MedDRA)[14] in study 3.
Statistical Analysis
Descriptive statistics were used to summarize the pharmacokinetic, tolerability and safety parameters. Pooled pharmacokinetic parameters were used to characterize the single-dose pharmacokinetics of armodafinil and its metabolites (R-modafinil acid and modafinil sulfone) across all three studies; multiple-dose parameters were obtained only in study 2. For study 3, in which each subject received armodafinil twice, the observations for each subject were averaged such that each subject within the study contributed only one observation to the pooled mean values following the administration of armodafinil. The summaries of combined pharmacokinetic parameters included all subjects with data that were adequate to fully characterize the pharmacokinetic profile using a noncompartmental approach.
Confirmation of steady state was evaluated via analysis of plasma concentrations of armodafinil normalized to a 50 mg dose on days 1, 7 and 14 of administration in subjects participating in the multiple-dose study. Attainment of steady state was further assessed for the 50, 100, 200, 250 and 300 mg doses by calculating the difference of log-transformed AUCτ and Cmax values between days 7 and 14 at each dose level via ANOVA, determining the mean difference of the log value and the 90% CI values at each dose, and then converting these values to geometric mean ratios and their 90% CIs. 90% CIs within the range 80–125% were taken as indicating equivalence of the day 7 and day 14 values at each dose and the attainment of steady state.
Dose-proportionality was assessed in study 2 by visual examination of the magnitude of increase in the mean values of AUC (AUC∞ on day 1; AUCτ on days 7 and 14) and Cmax relative to the magnitude of increase in armodafinil dose. In addition, the exposure-dose relationship was further evaluated using regression analyses of the log-transformed AUC and Cmax values as the dependent variable and the log value of dose as the independent variable on days 1, 7 and 14. Dose-proportionality was indicated when the 90% CI of the slope fell within the reference range of 0.8927–1.1073 for days 1 and 7 and 0.8755–1.1245 for day 14.[15]
Results
Of 256 subjects screened for participation in the three studies, 119 met the inclusion criteria and were randomized to receive armodafinil (either as a single dose or as multiple doses over 14 days; n = 97) or placebo (n = 22); 40 subjects were randomized in study 1, 49 in study 2, and 30 in study 3. All 119 subjects received at least one dose of study medication and composed the safety analysis set. The demographic features of the safety analysis set are shown in table I. The mean ages, bodyweight, height and BMI of subjects who received the various doses of armodafinil were similar to those of subjects who received placebo. The pharmacokinetic analysis set, which included all subjects in the safety analysis set who had at least one complete pharmacokinetic assessment, comprised 115 subjects (93 armodafinil, 22 placebo).

Demographic characteristics of the subjects who received at least one dose of armodafinil (range 50–400 mg) or placebo in the three studies
Of the 119 randomized subjects, 103 (85 armodafinil, 18 placebo) were considered ‘study completers’, having concluded all phases of their respective studies. 105 subjects (87 armodafinil, 18 placebo) completed all pharmacokinetic assessments and were considered ‘pharmacokinetic completers’; this number included two subjects who subsequently missed the final safety assessment. The 14 subjects who did not complete the pharmacokinetic assessments included three who were withdrawn because of adverse events, six in the 400 mg dose group in whom dosing was halted after day 7, one subject who withdrew consent, one who was a protocol violator, and three who were withdrawn for noncompliance or other reasons.
Pharmacokinetics of Armodafinil
Dose-Proportionality
Dose-proportionality of armodafinil was assessed across the range of 50–400 mg on days 1 and 7 and 50–300 mg on day 14 (table II). The pharmacokinetics, after both single and multiple doses, appeared to be linear, based on the approximately dose-proportional increases in AUC and Cmax over the dose ranges studied (figure 2 and figure 3). The slopes, calculated by linear regression analyses of log-transformed AUC and Cmax values as a function of log-transformed dose value, on days 1, 7 and 14 fell between 0.9 and 1.1 (table III). Although this pharmacokinetic study was not powered to support extensive statistical analysis, the 90% CIs of the slopes for AUCτ on day 7 and for Cmax and AUCτ on day 14 were found to fall within the corresponding reference ranges. The 90% CIs of the slopes for these parameters on day 1 and for Cmax on day 7 were only marginally outside the reference range.

Mean (± SD) maximum plasma concentrations (Cmax) and areas under the plasma concentration vs time curve (AUC) of armodafinil following single and multiple doses administered in the fasting state in the three studies

Mean (± standard error of the mean) armodafinil plasma concentrations on (a) day 1 and (b) day 7 in the multiple-dose study (n = 34) following doses of 50–400 mg in healthy men.

Individual and mean area under the concentration vs time curve (AUC) from time zero to infinity (AUC∞), AUC over one dosing interval (AUCτ), and maximum plasma concentration (Cmax) values of armodafinil following doses of 50, 100, 200, 250, 300 and 400 mg on (a) day 1 and (b) day 7 in the multiple-dose study in healthy men. The lines represent the best fit of the mean data and were obtained by linear regression analysis of AUC and Cmax data over the dose range.

Slopes and 90% CIs for dose-proportionality using log-transformed values of areas under the plasma concentration vs time curve (AUC) and maximum plasma concentrations (Cmax) as a function of dose following single and multiple doses in study 2
Mean Pharmacokinetic Parameters Following Single and Multiple Doses
Mean pharmacokinetic parameters of armodafinil following single and multiple doses derived from all three studies and normalized for a 50 mg dose are shown in table IV. These data confirm that armodafinil is readily absorbed after oral administration, and that Cmax is achieved approximately 2 hours after single- or multiple-dose administration in fasting subjects. After reaching peak, plasma concentrations declined in a relatively slow, apparently monophasic manner, with mean t½ β and CL/F values of approximately 14 hours and 39 mL/min, respectively, after single doses, and 17 hours and 33 mL/min, respectively, after 14 days of administration. The mean Vz/F was approximately 42 L after single doses and 47 L after 14 days of administration. These values approximate the estimated total body water in humans, indicating that armodafinil is widely distributed outside the vasculature, but does not appear to have high affinity for tissues.

Pharmacokinetic parameters for armodafinil and its metabolites R-modafinil acid and modafinil sulfone following single and multiple doses, normalized to a 50 mg dose of armodafinil (combined data)a
Plasma concentrations of armodafinil normalized to a 50 mg dose on days 7 and 14 in the multiple-dose study were similar, indicating that steady state had been attained within 7 days of once-daily administration (figure 4). Further confirmation of stable status was provided by the 90% CIs for geometric mean AUCτ ratios on day 7 versus day 14 for the armodafinil 50, 100, 200, 250 and 300 mg doses. For all five doses, the 90% CI bounds were well within the range 80–125% (data not shown). When this analysis was performed for Cmax, the 90% CI values were also well within the range 80–125% for all doses except the 250 mg dose (90% CI 74.9, 106.4 at this dose level).

Mean (± standard error of the mean) plasma concentrations of armodafinil normalized to a 50 mg dose over a 24-hour period on days 1, 7 and 14 of administration in the multiple-dose study in healthy men. The 400 mg dose was discontinued after day 7.
Plasma concentrations of armodafinil after multiple-dose administration were increased compared with those measured after single-dose administration (table IV), and the mean Robs was 1.8 on day 7 and 1.7 on day 14. The mean Rss was approximately 1.2 at both times, indicating that AUC values were only slightly larger on days 7 and 14 than would be predicted on the basis of single-dose data on day 1.
Influence of Food on Absorption
In the six subjects enrolled in study 1 who received single doses of armodafinil 100 mg both in the fasted state and after consumption of a fatty meal, absorption of the drug was slowed by the presence of food. Median tmax was approximately 4 hours longer when armodafinil was administered after a fatty meal compared with that following administration in the fasted state (6.0 vs 2.3 hours, respectively) [figure 5]. However, food had no effect on the overall bioavailability of armodafinil compared with fasting (Cmax 2.2 ± 0.1 vs 2.4 ± 0.4 µg/mL, respectively; AUC∞ 43.8 ± 8.2 vs 40.6 ± 7.4 µg • h/mL, respectively) [table V].

Mean (± standard error of the mean) plasma concentrations of armodafinil following doses of 100 mg in the fed and fasted states in the single-dose study in healthy men (n = 6).

Pharmacokinetic parameters of armodafinil following single 100 mg doses administered in the fasting state and after consumption of a standard fatty meal
Although the effect of food following multiple-dose administration was not studied, superimposition of the single-dose data indicated that a delay of approximately 2 hours in median tmax could be anticipated at steady state in the fed state compared with the fasted state (4.1 vs 1.9 hours, respectively).
Pharmacokinetics of R-Modafinil Acid and Modafinil Sulfone
The pharmacokinetic parameters determined for the two principal circulating metabolites of armodafinil are summarized in table IV. Systemic exposure to R-modafinil acid, as assessed by dose-normalized, mean AUC values, was approximately 11% of that of the parent drug after single-dose administration, and approximately 7% after multiple-dose administration. The median tmax and mean apparent t½ β values for this metabolite were similar to those of the parent drug. R-Modafinil acid achieved steady state within 7 days after initiation of dosing with armodafinil.
For modafinil sulfone, the dose-normalized mean AUC values (table IV) suggested that systemic exposure to this metabolite was approximately 33% of that of the parent drug after single-dose administration, and approximately 56% after multiple-dose administration. The mean t½ β values for modafinil sulfone were considerably higher than those of the parent drug, i.e. approximately 39 hours on day 14 versus 17 hours for armodafinil. Modafinil sulfone was not at steady state on day 7 but appeared to be at steady state by day 14.
Tolerability and Safety
Single doses of armodafinil were well tolerated at doses up to 400 mg. In subjects given multiple doses, doses up to 300 mg/day were well tolerated. The most frequent adverse events observed with armodafinil were headache, dizziness and nausea, which occurred in 42%, 22% and 19% of subjects, respectively (table VI); these adverse events appeared to be dose related, being more common with doses ≥250 mg. Most adverse events occurring with armodafinil were mild to moderate in nature and did not result in study withdrawal. The exception was the 400 mg group (multiple-dose), from which two subjects withdrew prior to the day 7 dose. Because of the high number of adverse events in the armodafinil-treated subjects in this group, dosing of the other six subjects scheduled to receive 400 mg, including those receiving matched placebo, was halted after day 7.

Adverse events [n (%)] occurring in ≥5% of subjects receiving armodafinil 50–400 mg or placebo (combined data)
No clinically meaningful changes in mean laboratory test values, physical examination or ECG findings were detected, and there were no overall trends suggesting a relationship to changes in mean vital sign parameters.
Discussion
Oral administration of modafinil results in significant differences in circulating concentrations of the two isomers of the drug, R and S, because of stereospecific differences in their elimination. For this reason, the plasma concentrations of R-modafinil (armodafinil) may be up to three times higher than those of S-modafinil.[9] Preclinical studies have shown that the isomers have approximately equipotent pharmacological effects.[8] This suggests that the majority of the effects observed with modafinil are attributable to the longer-lasting R-isomer. When given once daily in a previous study to healthy subjects who underwent acute sleep deprivation, single armodafinil 200 mg doses produced comparable Cmax values but higher concentrations later in the day (i.e. at 6–14 hours after administration) compared with modafinil 200 mg.[11] In addition, armodafinil 200 mg improved wakefulness and sustained attention for a longer period than modafinil 200 mg, as demonstrated objectively via the Maintenance of Wakefulness Test and Psychomotor Vigilance Task.[11] These findings suggest that because of the presence of the short-lived S-isomer in modafinil, the Cmax for modafinil would need to be substantially higher than that of armodafinil to maintain similar late-day exposure.
The present analysis involved pooling data from three separate, randomized investigations of armodafinil in healthy volunteers in order to assess the single- and multiple-dose pharmacokinetics and safety/tolerability of armodafinil and its two circulating metabolites, R-modafinil acid and modafinil sulfone. Armodafinil displayed linear pharmacokinetics over the 50–400 mg dose range, with Cmax occurring approximately 2 hours after administration in fasting subjects. Dose-proportionality at steady state was confirmed by linear regression analyses of the log-transformed AUC and Cmax values as a function of dose in the multiple-dose study. Compared with the biphasic decline from peak shown by modafinil,[9,10] plasma concentrations of armodafinil declined from peak in a monophasic manner, with a mean t½ β of approximately 15 hours. When assessed in subjects who had eaten a fatty meal, tmax occurred later, but systemic exposure (Cmax and AUC) appeared to be only minimally affected, suggesting that the presence of food may slow the rate of absorption of armodafinil without materially affecting the extent of absorption.
The results of the current analysis are consistent with those of an earlier study by Dinges et al.,[11] in which single oral doses of armodafinil in the range 100–300 mg were administered to 71 healthy male volunteers aged 18–40 years. Armodafinil displayed linear pharmacokinetics over this dose range, as it did in the present analysis, and Cmax values were comparable to those noted in our investigation. Because subjects in the study by Dinges et al.[11] received armodafinil doses immediately followed by a standardized meal, tmax occurred later than those reported for subjects who had fasted (5–6.5 hours vs 1.5 hours), but were comparable to those found in subjects who received an armodafinil dose of 100 mg after a fatty meal in this analysis.
The results of the present analysis are also consistent with those previously published for armodafinil when plasma samples were analysed using chiral bioanalytical methods after administration of modafinil. In healthy male subjects who received modafinil 400 mg once daily for 7 days,[9] pharmacokinetic parameters for armodafinil were similar to those in subjects who received armodafinil 200 mg once daily in the current multiple-dose study. On day 7, R-modafinil Cmax, CL/F and AUCτ values were 6.8 µg/mL, 34.0 mL/min and 104.0 µg • h/mL, respectively, after administration of modafinil 400 mg in the study by Wong et al.[9] compared with 7.4 µg/mL, 32.4 mL/min and 111.8 µg • h/mL after armodafinil 200 mg in the current multiple-dose study. The similarity of the results for armodafinil after administration with those obtained after administration of modafinil suggests that there is little, if any, pharmacokinetic interaction between the isomers in humans.
Analysis of the pooled safety data from the three randomized studies showed that armodafinil was generally well tolerated after single and multiple doses, particularly at doses <250 mg/day, in healthy subjects. The most frequent adverse events noted with armodafinil were headache, dizziness and nausea, which appeared to be dose-dependent.
Conclusions
In the present analysis, armodafinil displayed linear pharmacokinetics over the dose range of 50–400 mg. Cmax was reached in approximately 2 hours when the drug was taken on an empty stomach; food affected the rate but not the extent of absorption. Steady state appeared to be reached within 7 days after commencing once-daily administration. After reaching Cmax, concentrations of armodafinil exhibited an apparent monophasic decline, with a mean t½ β of around 15 hours. Armodafinil was generally well tolerated in these healthy volunteers; adverse events were dependent upon dose.
References
Roth T, White D, Schmidt-Nowara W, et al. Effects of armodafinil in the treatment of residual excessive sleepiness associated with obstructive sleep apnea/hypopnea syndrome: a 12-week, multicenter, double-blind, randomized, placebo-controlled study in nCPAP-adherent adults. Clin Ther 2006; 28(5): 689–706
Hirshkowitz M, Black JE, Wesnes K, et al. Adjunct armodafinil improves wakefulness and memory in obstructive sleep apnea/ hypopnea syndrome. Respir Med 2007; 101(3): 616–27
Drake C, Walsh J, Roth T. Armodafinil improves sleep latency in patients with shift work sleep disorder [abstract no. 0189]. Sleep 2006; 29 Suppl.: A64
Harsh JR, Hayduk R, Rosenberg R, et al. The efficacy and safety of armodafinil as treatment for adults with excessive sleepiness associated with narcolepsy. Curr Med Res Opin 2006; 22(4): 761–74
Schwartz JR, Nelson MT, Schwartz ER, et al. Effects of modafinil on wakefulness and executive function in patients with narcolepsy experiencing late-day sleepiness. Clin Neuropharmacol 2004; 27(2): 74–9
Schwartz JR, Feldman NT, Bogan RK, et al. Dosing regimen effects of modafinil for improving daytime wakefulness in patients with narcolepsy. Clin Neuropharmacol 2003; 26(5): 252–7
Schwartz JR, Feldman NT, Bogan RK. Dose effects of modafinil in sustaining wakefulness in narcolepsy patients with residual evening sleepiness. J Neuropsychiatry Clin Neurosci 2005; 17(3): 405–12
Data on file, Cephalon, Inc., West Chester (PA), USA, 1998
Wong YN, Simcoe D, Hartman LN, et al. A double-blind, placebo-controlled, ascending-dose evaluation of the pharmacokinetics and tolerability of modafinil tablets in healthy male volunteers. J Clin Pharmacol 1999; 39(1): 30–40
Wong YN, King SP, Simcoe D, et al. Open-label, single-dose pharmacokinetic study of modafinil tablets: influence of age and gender in normal subjects. J Clin Pharmacol 1999; 39(3): 281–8
Dinges DF, Arora S, Darwish M, et al. Pharmacodynamic effects on alertness of single doses of armodafinil in healthy subjects during a nocturnal period of acute sleep loss. Curr Med Res Opin 2006; 22(1): 159–67
Expert Working Group (Efficacy) of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Good Clinical Practice: Consolidated Guidance E6(R1). ICH, 1996 [online]. Available from URL: http://http://www.fda.gov/cder/guidance/959fnl.pdf [Accessed 2008 Nov 25]
United States Food and Drug Administration, Center for Drug Evaluation and Research. Coding symbols for thesaurus of adverse reaction terms. 5th ed. Rockville (MD): Department of Health and Human Services, Center for Drug Evaluation and Research, Office of Epidemiology Biostatistics, 1995
International Federation of Pharmaceutical Manufacturers Associations. Medical dictionary for regulatory activities (Med-DRA): version 7.0. Chantilly (VA): MedDRA MSSO, 2004 Mar
Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res 2000; 17(10): 1278–83
Acknowledgements
Cephalon, Inc., Frazer, PA, USA, sponsored this pooled analysis and, with guidance from the authors, was responsible for the design of the study and data analysis. The authors were employees of Cephalon, Inc. at the time of the analyses and manuscript development. The authors wish to acknowledge the study investigators who enrolled subjects and collected data under the direction of the sponsor: Stephen Freestone (Inveresk Research [now known as Charles River Laboratories], Tranent, Scotland, UK; studies 1 and 2), and Dennis Swearingen (MDS Pharma Services, Phoenix, AZ, USA; study 3). John G. Jiang, a former employee of Cephalon, Inc., contributed to the analysis of the data. Virginia Schobel, an employee of Cephalon, Inc., and Thomson Reuters (Horsham, PA, USA) provided editorial assistance to the authors. All authors had full access to the data and contributed to data interpretation and preparation of this report. The corresponding author had final responsibility for the decision to submit this report for publication.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Darwish, M., Kirby, M., Hellriegel, E.T. et al. Pharmacokinetic Profile of Armodafinil in Healthy Subjects. Clin. Drug Investig. 29, 87–100 (2009). https://doi.org/10.2165/0044011-200929020-00003
Published:
Issue Date:
DOI: https://doi.org/10.2165/0044011-200929020-00003