Article Text

Abstract
Tuberculosis remains a major health problem, with two million deaths and eight million new cases annually. At the same time, two billion people (one third of the total world population) are infected with the aetiological agent, Mycobacterium tuberculosis. Of these, fewer than 10% ever develop disease, although the pathogen is not eradicated but rather contained in discrete lesions. Hence, the immune system is highly effective in containing the pathogen, but fails to eradicate it. Disease typically develops through reactivation once the immune system is weakened. The immune response to M tuberculosis is T cell dependent. It comprises not only the conventional CD4 and CD8 T cells, but also γδ T cells and CD1 restricted T cells. γδ T cells recognise phospholigands and no presentation molecules are known thus far. CD1 restricted T cells recognise glycolipids, which are highly abundant components of the mycobacterial cell wall. Although different T cells are required for optimum protection, the immune mechanisms known to have a role in acquired resistance can be associated with two major mechanisms: (a) activation of macrophages by cytokines; (b) direct cytolytic activity. In vivo granuloma formation, which is central to protection, is induced and sustained by cytokines. Mycobacteria are contained within granulomas and in this way are prevented from spreading all over the body.
- tuberculosis
- cytokines
- T cells
- macrophages
- HIV, human immunodeficiency virus
- IFNγ, interferon γ
- LTα, lymphotoxin α
- MDR, multidrug resistant
- MHC, major histocompatibility complex
- TNFα, tumour necrosis factor α
Statistics from Altmetric.com
- HIV, human immunodeficiency virus
- IFNγ, interferon γ
- LTα, lymphotoxin α
- MDR, multidrug resistant
- MHC, major histocompatibility complex
- TNFα, tumour necrosis factor α
Tuberculosis still remains a major health problem globally.1 In 2000, 1.7 million people died of this disease (table 1). These are 3% of all premature deaths that occurred in that year—more than diabetes mellitus (1.4%), congenital abnormalities (1.2%), Alzheimer’s disease (0.5%), and rheumatoid arthritis (0.01%) together. Also, the number of life years lost owing to tuberculosis is tremendous: 33.3 million man years (2.3% of all losses), more than the man years lost due to diabetes mellitus (1%), Alzheimer’s disease (0.7%), and rheumatoid arthritis (0.3%) together. The figures are even more dramatic if we consider the dangerous liaison between tuberculosis and the acquired immune deficiency syndrome (AIDS). Half a million people died in the past year because of co-infection with Mycobacterium tuberculosis and the human immunodeficiency virus (HIV). The problem is further complicated by the increasing numbers of multidrug resistant (MDR) strains. More than 50 million people are already infected with MDR strains of M tuberculosis, and in many states more than 15% of all tuberculosis cases are caused by MDR strains. This enormously complicates the difficult treatment schedules and increases the cost of treatment from $100 to $100 000 in industrialised countries. In many developing countries, MDR tuberculosis can practically no longer be cured. Prisons and work camps in Russia are major breeding centres of MDR tuberculosis.
Tuberculosis: still a major global threat as we have entered the new millennium
THE DISEASE AND THE UNDERLYING IMMUNE RESPONSE
Tuberculosis typically is a disease of the lung, which serves both as port of entry and as the major site of disease outbreak.2 Inhaled droplets containing minute numbers of bacteria are engulfed by alveolar macrophages. These macrophages harbour the pathogen and transport it to draining lymph nodes. A small granulomatous lesion develops containing the bacteria (fig 1). This is true for about 90% of all of those who are infected. They will not develop disease directly. Still, because the bacteria are not eradicated, the risk remains that disease may develop later. Although we cannot formally exclude the possibility of spontaneous healing promptly after infection with M tuberculosis, this possibility seems extremely unlikely. In people with a compromised immune system, however, disease will develop directly after primary infection. This, for example, is the case in patients with HIV infection, in whom the risk of disease development in the following year is dramatically increased depending on the severity of immunodeficiency. Those who contain the infection for years remain at risk and at a later time, when the immune system is weakened, outbreak may occur owing to reactivation. Although considered less likely, reinfection leading to disease is not excluded. Bacterial containment is focused on the granulomatous lesion, where different T cell populations participate in the protective immune response. These include the following T cell populations:
CD4 T cells recognising antigenic peptides in the context of gene products encoded by the major histocompatibility complex (MHC) class II
CD8 T cells recognising antigenic peptides in the context of MHC class I
γδ T cells recognising unusual antigenic ligands independently of specialised presentation molecules—notably, phospholigands
CD1 restricted T cells recognising glycolipids abundant in the mycobacterial cell walls presented by the CD1 molecules.

Different outcomes of infection with M tuberculosis, different T cell populations involved in protection, and major anti-mycobacterial effector mechanisms of macrophages. This scheme firstly depicts the different outcomes of tuberculosis in healthy and immunocompromised subjects. Secondly, the figure shows the different T cell populations and their major T cell effector mechanisms in the control of disease. Thirdly, the figure shows anti-mycobacterial effector mechanisms of activated macrophages. (Reproduced from Nature Reviews Immunology (vol 1:20–30). Reprinted by permission from Nature Reviews Immunology (2001;1:20–30). Copyright © 2001 Macmillan Magazines Ltd.)
MYCOBACTERIA, MACROPHAGES, CYTOKINES, AND T LYMPHOCYTES
M tuberculosis is an intracellular pathogen with the ability to persist in the early phagosomal compartment.2 It arrests phagosome maturation at an early stage and strongly inhibits phagolysosome fusion. The phagolysosome is a rather hostile environment, where many bacterial pathogens are killed. The early phagosome is a less hostile compartment where M tuberculosis can accommodate itself. Yet, arrest of phagosome maturation by M tuberculosis is not complete and some bacteria are killed or at least prohibited from replication through antibacterial mechanisms including reactive oxygen and nitrogen intermediates, which are produced by activated macrophages (fig 1). The different T cell populations produce interferon γ (IFNγ) and hence are of the T helper 1 (Th1) type. This cytokine is the central mediator of macrophage activation. IFNγ synergises with tumour necrosis factor α (TNFα) in activating macrophages. CD4 T cells also produce lymphotoxin α (LTα), which participates in protection against tuberculosis. At least some of the CD8 T cells, γδ T cells, and CD1 restricted T cells secrete perforin and granulysin which directly kill mycobacteria within macrophages (fig 1).
The costimulatory activity of TNFα in IFNγ activation of anti-mycobacterial macrophage functions was established in the early 1990s. In a series of experiments, Flesch and Kaufmann showed that IFNγ rather than TNFα stimulates anti-mycobacterial macrophage functions in vitro.3 However, blocking of endogenously produced TNFα reduced IFNγ induced macrophage activation and combination of both TNFα and IFNγ induced optimum macrophage activation. In subsequent studies, the role of TNFα in protection against tuberculosis was elucidated. Gene knockout mice deficient in TNFRI have exacerbated tuberculosis.4 In particular, granuloma formation in these gene knockout mice is impaired. Moreover, it was found that neutralisation of TNFα in mice with latent tuberculosis results in reactivation and outbreak of disease.5 These data provide convincing evidence that in vivo TNFα is mostly involved in bacterial containment and in the formation and sustainment of granulomatous lesions. The availability of mice deficient in LTα and LTβ showed that in addition to TNFα, LTα participates in granuloma formation.6 Hence LTα and TNFα both contribute to granuloma formation, but they do so independently of each other. Figure 2 shows the development of a tuberculous lesion as well as the T cell subsets and cytokines operative in the lesion.

Development of granulomatous lesions in tuberculosis. Promptly after infection, T cells and macrophages are attracted to the site of mycobacterial implantation. There, granulomatous lesions develop. As long as the immune response is competent, the lesions will contain bacteria. These productive granulomas represent a focus of highly dynamic interactions between different T cell populations, macrophages of different maturation stages, and dendritic cells. Once immunity weakens, the balance is tipped and the granuloma can no longer contain mycobacteria. Rather, the granulomatous lesion liquefies and bacteria are released to different tissue sites, different organs, and to the environment. Active disease develops and the patient becomes contagious. (For further details see ref 8.)
T LYMPHOCYTE POPULATIONS AS CENTRAL MEDIATORS OF PROTECTION
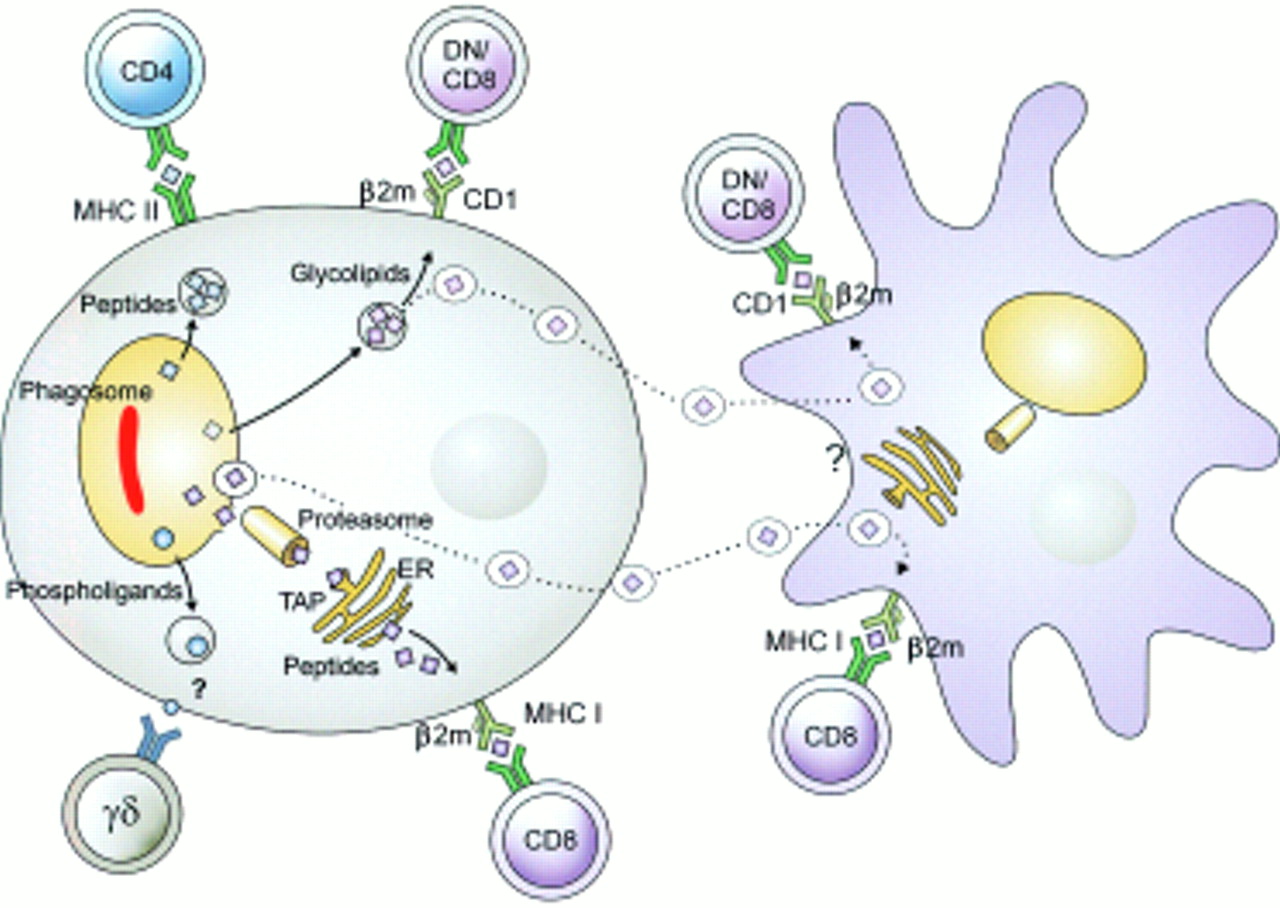
Because M tuberculosis resides in the phagosomal compartment, its proteins have ready access to the MHC class II processing and presentation machinery.2 In contrast, it is still incompletely understood how mycobacterial antigens gain access to the MHC class I processing machinery. Similarly, the question of how mycobacterial glycolipids are presented by CD1 molecules remains incompletely understood. CD1b, a major presentation molecule of this pathway, is found in the late phagosomal compartment, which is reached by only very few M tuberculosis organisms (see above). Moreover, macrophages, which are the major habitat of M tuberculosis are virtually devoid of surface expressed CD1 molecules and hence fail to present mycobacterial glycolipids. Some intracellular bacteria, such as Listeria monocytogenes, egress from the phagosomal compartment into the cytoplasm. In the cytoplasm, their antigens have ready access to MHC class I processing. It has been proposed that M tuberculosis can also form pores in the phagosomal membrane. Because M tuberculosis apparently remains in the phagosome, it was furthermore assumed that these pores allow exchange of molecules between the phagosome and the cytoplasm. This would not only provide cytoplasmic nutrients to M tuberculosis but also promote antigen translocation to the MHC class I pathway. More recent studies, however, have produced evidence arguing against translocation of phagosomal molecules into the cytoplasm and, as a corollary, against direct antigen transport to MCH class I (Schaible UE, et al, unpublished data). However, M tuberculosis is a potent inducer of apoptosis in host macrophages, which leads to the production of extracellular vesicles carrying a variety of antigens including glycolipids and proteins. These extracellular vesicles are readily taken up by dendritic cells, which then present their antigenic cargo in the context of MHC I and CD1. Subsequently, CD8 T cells and CD1 restricted T cells are stimulated. This hypothesis accounts for T cell stimulation through MHC class I and CD1 despite macrophage deactivation, deficient CD1 expression by macrophages, and lack of pore formation in phagosomal membranes during tuberculosis. Hence, this hypothesis accounts for several thus far unresolved obstacles of T cell activation during tuberculosis. Figure 3 summarises this hypothesis.
{kind=link}
{kind=link}
{kind=link}
Different antigen presentation pathways in tuberculosis. Mycobacterial antigens reside in the early phagosome. There, their proteins have ready access to MHC class II processing, resulting in potent CD4 T cell stimulation. Phospholigands are produced by these mycobacteria, which stimulate γδ T cells in the absence of known antigen presentation molecules. Presentation of proteins by MHC class I and of glycolipids by CD1 is more complex and probably requires cross priming. Mycobacteria infected macrophages undergo apoptosis. Resulting extracellular vesicles carry antigens to bystander dendritic cells. Uptake of these vesicles results in glycolipid presentation through CD1 and protein presentation through MHC class I. This two cell mechanism can explain stimulation of MHC class I restricted CD8 T cells and of CD1 restricted T cells. (Reproduced from Nature Reviews Immunology (vol 1:20–30). Reprinted by permission from Nature Reviews Immunology (2001;1:20–30). Copyright © 2001 Macmillan Magazines Ltd.)
CONCLUDING REMARKS
Tuberculosis is a chronic disease with protection strongly depending on mycobacterial containment to discrete granulomatous lesions. The mechanisms underlying this containment are highly complex comprising different T cell populations. Although CD4 T cells, CD8 T cells, γδ T cells, and CD1 restricted T cells are apparently all required for optimum protection, a hierarchy seems to exist: CD4 T cells are the most important players, followed by CD8 T cells. The role of these T cell populations in protection against tuberculosis is well understood because of the availability of suitable animal models. The roles of γδ T cells and CD1 restricted T cells are less well understood as appropriate animal models are not available. It is, however, fair to assume that these T cell populations also participate in protective immunity. Although T cells are the major mediators of protection, effector functions are mostly provided by macrophages. The cross talk between T cells and macrophages is achieved by various cytokines, notably IFNγ, TNFα, and LTα. This cross talk first results in macrophage activation—that is, the ability of macrophages to control mycobacterial replication. Secondly, this cross talk causes the formation of well organised granulomas, where macrophages, dendritic cells, and different T cell populations exist in near vicinity. As long as this cross talk is well balanced, productive granulomas develop which contain the bacterium. Once any element of this complex interplay is impaired, the balance is tipped and the productive granuloma can no longer be sustained. Caseous lesions develop which fail to contain the bacteria. Consequently, bacteria can be disseminated to other tissue sites, to other organs, and even to the environment: active disease develops and the patient becomes contagious. Experiments with gene knockout mice with TNFα signalling defects have already pointed to a central role for this cytokine in the maintenance of granulomatous lesions and control of disease.4 Patients with a hereditary defect in TNFα signalling and with tuberculosis have not been described. Therefore, activation of tuberculosis in patients with rheumatoid arthritis treated with anti-TNFα antibodies underline the importance of this cytokine in the control of latent human tuberculosis.7
Acknowledgments
The superb help of Mrs Sibaei is gratefully acknowledged. The author’s work on tuberculosis receives financial support from BMBF, EC, DFG, WHO GPV-VRD, and Fonds Chemie.