Article Text

Abstract
An increase in the frequency of diagnosing non-cystic fibrosis bronchiectasis in children is due to heightened awareness of the disease and the wider availability of high-resolution computed tomography. The most common underlying conditions leading to bronchiectasis include infections, immunodeficiency, aspiration and primary ciliary dyskinesia. Treatment centres on airway clearance with aggressive antibiotic regimens and physiotherapy; more specific approaches are available for some of the underlying conditions. A high index of suspicion that a child may have underlying bronchiectasis must be maintained in the presence of prolonged or recurrent “wet/productive” cough. The classic definition of bronchiectasis is of irreversible bronchial dilatation; however, at the milder end of the spectrum it appears that radiographic changes may be reversible. Untreated, in its severest form bronchiectasis can progress to end stage pulmonary failure in adult life. In this article, we review its pathogenesis and diagnosis and the evidence base for available treatments.
Statistics from Altmetric.com
Introduction
Bronchiectasis, although probably first noted by Jean-Bruno Cayol in 1808,1 was first described by Rene Laënnec, the man who invented the stethoscope, in 1819.2 Sir William Osler presented a more thorough differentiation in the late 19th century and it is suspected that he actually died from complications of undiagnosed bronchiectasis.3 The term bronchiectasis originates from the Greek and translates as “stretching of the wind pipe”, a reference to abnormally dilated and often thick walled bronchi.
Less than 20 years ago non-cystic fibrosis bronchiectasis was termed an “orphan disease”4 because of the perception that it was a rare occurrence in the developed world. Population data are scarce and epidemiological evaluation is hampered by varying denominators. In Finland, one of the few developed countries publishing national data, the annual incidence has been estimated at 0.5 per 100 000 children under the age of 15 years.5 The highest prevalence reported worldwide appears to be among indigenous communities such as Southwest Alaskan Native children, with an annual incidence of 16:1000,6 and Central Australian Aborigine children, with an annual incidence of15:1000.7 8 Genetic factors may play a role as suggested by data from a study from New Zealand where annual incidence varied between 1.5 and 17.8 per 100 000 children between different ethnic groups.9
Bronchiectasis appears to be more common among children in developing countries and in lower socioeconomic classes.10 This is thought to be a result of more frequent respiratory infections, environmental airway irritants, poor immunisation rates and malnutrition. More recently, however, and with the advent of more sophisticated diagnostic tools such as high-resolution computed tomography (HRCT), a greater number of children are being diagnosed in the developed world. A large number of these children present with early disease and prompt recognition in combination with aggressive treatment have led to reconsideration of the traditional concept of irreversible lung damage.11
Pathophysiology
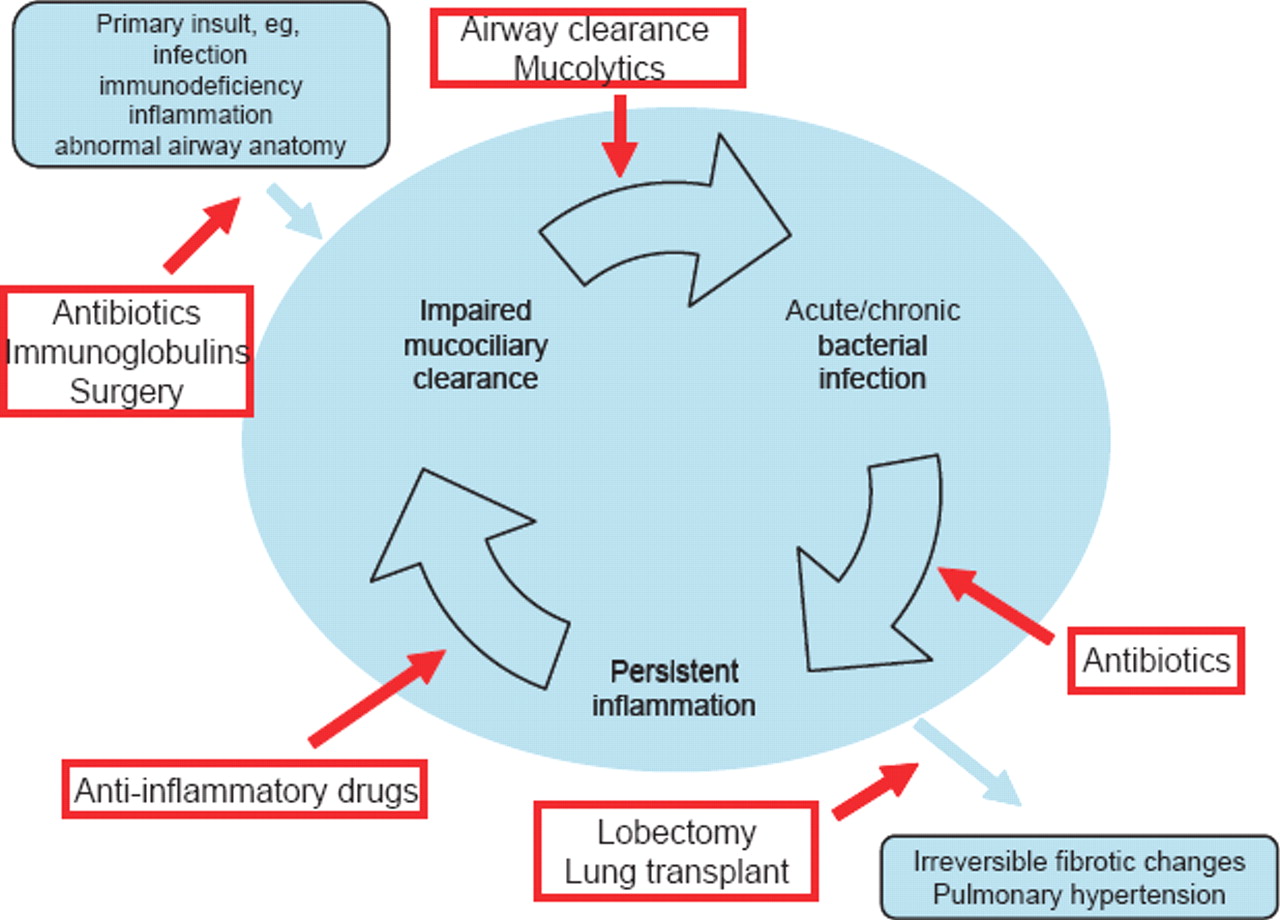
Several mechanisms have been proposed for the development of bronchiectasis, all linked to the different aetiologies associated with it. The most likely chain of events starts with obstruction of the bronchial tree, usually caused by infection and inflammation. The host response, especially after repeated insult, will then cause destruction of the bronchial ciliated epithelium. This leads to impaired mucociliary clearance and retention of secretions, setting in motion a vicious circle with ensuing chronic bacterial infection and persistent inflammatory response (figure 1). Histologically, damaged ciliated epithelium can be regenerated as cuboidal and later on as squamous epithelium. Even in the early stages, surrounding parenchyma is infiltrated by inflammatory cells. Further destruction of neighbouring tissue is seen if the disease progresses to cylindrical and saccular forms of bronchiectasis, with damage extending to muscle layers, surrounding cartilage and vascular structures, causing obstructive endarteritis and eventually pulmonary hypertension at the severest end of the spectrum.12,–,14

Pathogenesis and interventions.
Aetiology
Although in a large number of cases an underlying cause cannot be identified, a wide range of conditions can give rise to non-cystic fibrosis bronchiectasis (box 1).15 16 Familiarity with the most common and treatable causes is important since retrospective studies have reported a delay of 311 to 517 years between onset of respiratory symptoms and diagnosis, leaving the bronchial tree open to ongoing damage.
Box 1 Aetiology of childhood non-cystic fibrosis bronchiectasis1516
Post infectious disease*
Bacterial (Bordetella pertussis, necrotising bacterial pneumonia: staphylococcal, Klebsiella, pseudomonal)
Viral (measles, adenovirus, respiratory syncitial virus)
Other (tuberculosis, Apergillus fumigatus, Mycoplasma pneumoniae)
Congenital
Structural (congenital lobar emphysema, airway malacia, Williams–Campbell syndrome, tracheomegaly, Marfan syndrome)
Other (yellow nail syndrome, α-I antitrypsin deficiency)
Congenital immunodeficiencies*
Hypogammaglobulinaemia (panhypogammaglobulinaemia, eg, X-linked agammaglobulinaemia, severe combined, common variable immunodeficiency)
Functional antibody deficiency
Complement pathway defects (mannose binding ligand)
Neutrophil function abnormalities (Schwachman–Diamond syndrome, chronic granulomatous disease, Wiskott Aldrich syndrome)
MCH class 2 deficiency
Acquired immunodeficiencies
HIV infection
Drug induced (post chemo-/radiotherapy, steroids)
Ciliary abnormalities* (Kartagener syndrome, primary ciliary dyskinesia)
Mechanical*
Foreign body aspiration
Extrinsic compression (pulmonary artery sling, bronchocoele)
Endobronchial lesion (tumour)
Aspiration* (single episode or chronic)
Gastro-oesophageal reflux
Swallowing dysfunction
Tracheo-oesophageal fistula
Laryngeal cleft
Other
Inhalation of toxic gases
Idiopathic
↵* Indicates most commonly identified causes.
Post infectious disease
Historically, severe pulmonary infections were thought to account for the majority of cases of non-cystic fibrosis bronchiectasis.18 19 Although as a result of better sanitation, early immunisation and generous use of antibiotics (especially in the developed world), intrinsic causes are becoming more important,20 recent case series suggest that post infectious bronchial damage still accounts for the majority of cases.15 21 The relative frequency of causative organisms has changed and while measles, pertussis and tuberculosis are becoming rarer, recurrent viral infections are probably on the rise.22 Viruses cause distortion of airway surface morphology and disrupt cilial function for many weeks,23 causing bacteria to persist in the airways and trigger the inflammatory circle. Bronchiectasis can also result from bronchiolitis obliterans, regardless of its aetiology.24 25
Immunodeficiencies
Congenital and acquired immunodeficiencies account for between 18%17 and 34%15 of childhood cases of bronchiectasis. They cause increased susceptibility to a range of infections including the common respiratory pathogens and those that may not commonly threaten the immunocompetent host. In some conditions, such as common variable immunodeficiency (CVID), antibody response to infection can change over time, so that immunoglobulins and vaccine antibodies may have to be repeatedly checked in light of clinical concern. Immunoglobulins are often low in infants as a result of maturational delay and these patients may require supplementation with intravenous or subcutaneous human immunoglobulin to prevent progression to bronchiectasis. The identification of an organism is not only important for treatment guidance but can also be a useful pointer for detection of the underlying condition, for example, cytomegalovirus and Pneumocystis jirovecii in T cell defects, staphylococci in chronic granulomatous disease and Herpes viridae in disorders affecting natural killer cells.26
Ciliary abnormalities
The proportion of non-cystic fibrosis bronchiectasis cases due to primary ciliary dyskinesia (PCD) varies between 1.5% and 15% according to case series. PCD presents with a combination of upper and lower respiratory tract symptoms caused by inefficient mucociliary clearance. In view of the high frequency of these symptoms in childhood, the need for an increased index of suspicion in general paediatric clinics has been pointed out.27 Mirror image arrangement (figure 2) is thought to be present in nearly 50% of PCD patients,28 although the majority of patients in a PCD clinic might be affected owing to under-diagnosis of those with normal situs. Kartagener syndrome consists of bronchiectasis, sinusitis and situs invertus and with appropriate treatment regimens it might be possible to avoid progression to bronchiectasis, hence reducing the proportion of PCD patients affected. Nasal and exhaled nitric oxide (NO) are abnormally low29 30 and nasal NO can be used as a screening test31 in co-operative children. There is an overlap with other inflammatory lung diseases such as cystic fibrosis that also produce a low upper airway NO32 and the diagnosis rests on cilia examination with light and electron microscopy.

High-resolution computed tomography (HRCT) – situs inversus in a child with primary ciliary dyskinesia and associated bronchiectasis.
Mechanical causes
In a retrospective case review of 654 children undergoing flexible bronchoscopy in Istanbul, foreign bodies were identified in 4.8% with a median age of 2.5 years and a median symptomatic period of 3 months. In none of the patients was a history of foreign body aspiration forthcoming. The most common misdiagnosis was bronchitis.33 A long-term follow-up study performed by the same group showed an increased risk of complications associated with increasing elapsed time from aspiration to diagnosis. Bronchiectasis was found in 25% of patients whose diagnosis was delayed by more than 30 days.34 Rarer mechanical causes include bronchial obstruction by other thoracic structures such as pulmonary artery slings, bronchioceles or tumours.
Aspiration
Gastro-oesophageal reflux with silent aspiration is also thought to be a cause of bronchiectasis, especially in the neurologically impaired with poor swallow reflexes. A series of chest radiographs showing abnormalities in different lobes at different times should arouse suspicion. Changes are usually present in both lower lobes and there can be a predilection for the “dependent” lobe—the right upper in infants (figure 3) or children who spend significant amounts of time in a supine position, or the superior branch of the right lower lobe in the older child.
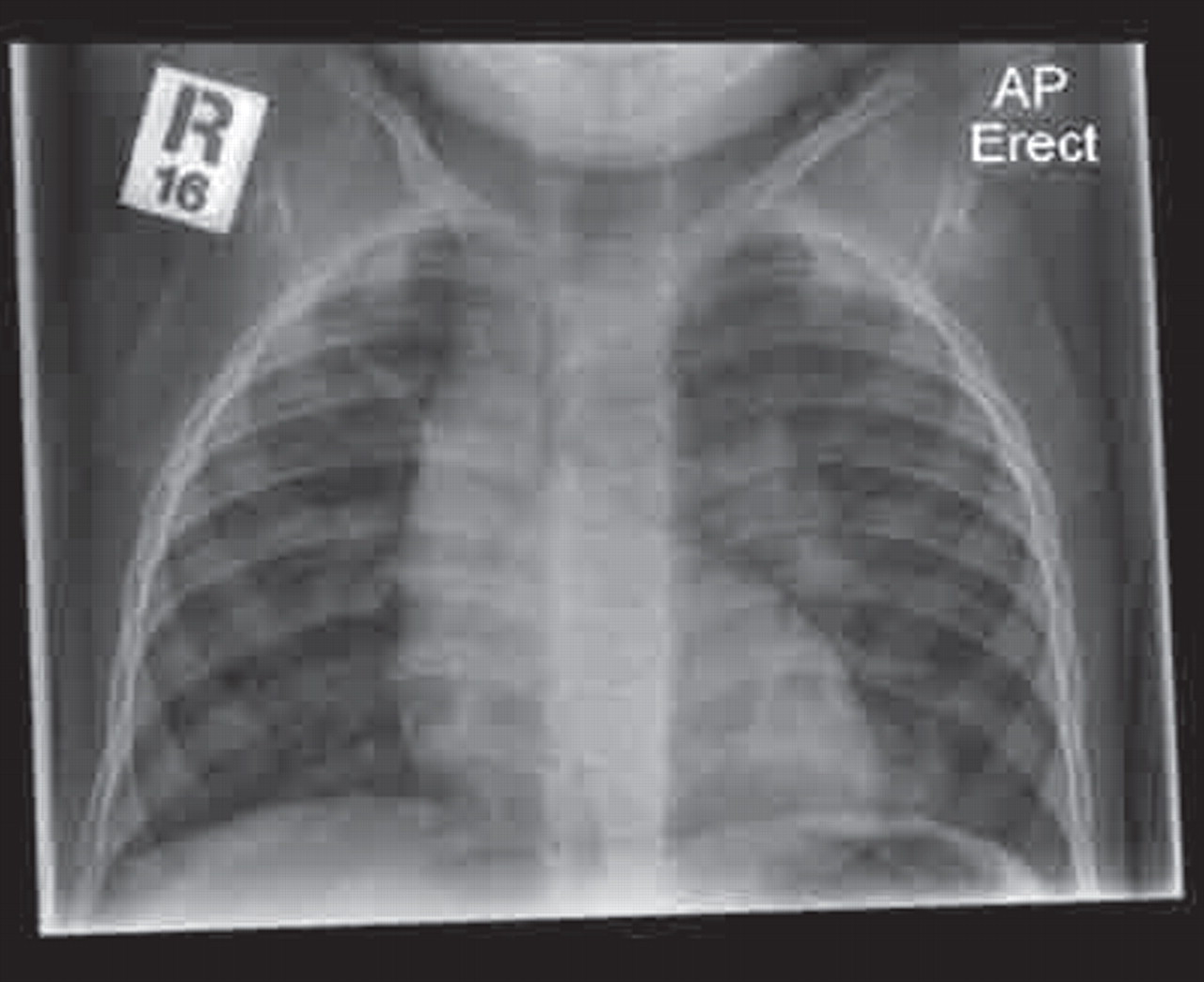
Right upper lobe collapse in an infant with severe gastro-oesophageal reflux (horizontal fissure pulled up from collapse).
Laryngeal clefts and tracheo-oesophageal fistulas are associated with primary (from “above”) as well as secondary (from “below”) aspiration and can hence give rise to bronchiectasis,35 particularly when compounded by dysphagia as is often the case in this group of patients.
Non-classical cystic fibrosis
A group of patients with suppurative lung disease and borderline sweat chloride results (30–60 mmol/l) previously regarded as having non-cystic fibrosis bronchiectasis are now more commonly classified as “non-classical cystic fibrosis”. This new diagnostic approach considers genetic, molecular, biological and clinical manifestations separately36 and makes allowances for the wide variety of phenotypic presentations ranging from male infertility37 to classical disease with a completely normal cystic fibrosis gene sequence.38 39
Making the diagnosis
Chronic cough
The assessment and management of cough in children was revised in a recent British Thoracic Society guideline40 which suggests that cough lasting for 8 weeks or longer ought to be considered for investigation with a chest radiograph.
The key to a raised level of suspicion in chronic cough is the presence of a “wet” cough, suggesting the production of sputum which children often do not expectorate.41 Studies into chronic cough have highlighted the importance of identifying and treating persistent bacterial bronchitis in view of the risk of progression to bronchiectasis.42 It is important to note in this context that many children with chronic cough and no evidence of reversible airway disease or atopy are misdiagnosed with asthma, in particular “cough variant asthma”, an entity which might not actually exist.41 The truly asthmatic child usually displays a dry and tight rather than wet cough. In the presence of a wet cough, asthmatic patients ought to be investigated for co-existing or alternative conditions.
Chest radiograph
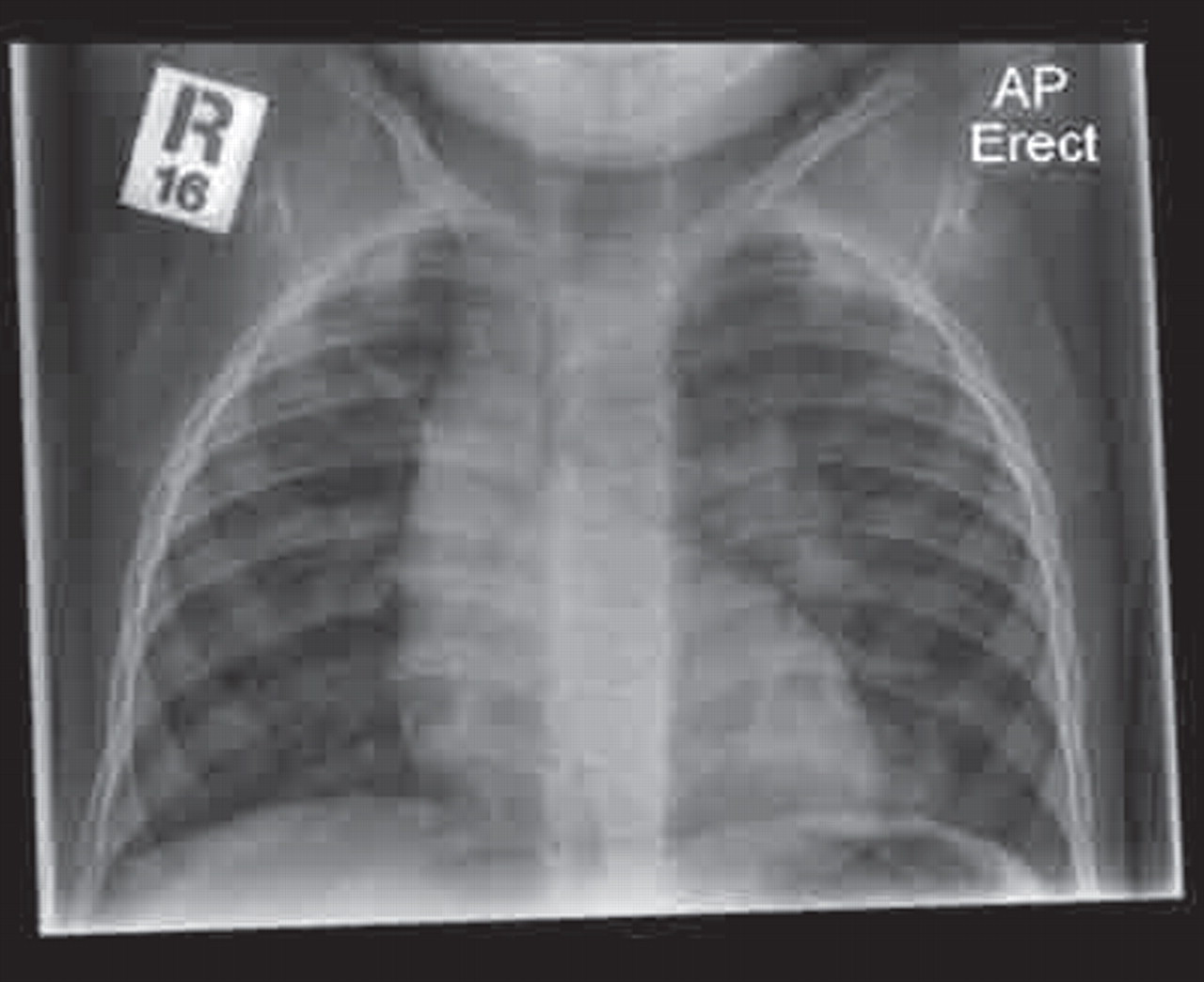
A plain chest radiograph is essential and may arouse suspicion with increased peribronchial markings or even obvious areas of damage (figure 4). It will give a useful overview of the general state of the lungs, but bronchiectasis cannot reliably be excluded and a suggestive clinical history should prompt further investigation with chest CT even in the absence of obvious changes on a chest radiograph. Plain chest radiograph changes are more easily visible in established bronchiectasis and include parallel line opacities (tramlining; see figure 5) caused by thickened dilated bronchi.

Chest x-ray showing evidence of likely bronchiectasis adjacent to right cardiac border (arrow).

Plain chest x-ray showing evidence of tramlining in left lower lobe (arrow).
High-resolution computed tomography
HRCT is now widely available and recognised as the gold standard for diagnosis. Tracheobronchograms, which were previously used as a diagnostic tool, are less used now because of technical difficulties in their performance, the need for general anaesthesia and post procedural atelectasis.41 42 Despite improvements in imaging technology resulting in a reduced scanning time of a few minutes, it is essential for the quality of the images that the child lies still. Infants can sometimes be fed and wrapped, although expiratory films that are useful when looking for air trapping will then not be possible. Toddlers and young children may require sedation or short general anaesthesia to obtain good quality films.
The widely accepted Naidich criteria for radiological bronchiectasis require demonstration of a lack of normal bronchial tapering or any bronchi with an internal diameter greater than the diameter of the accompanying pulmonary artery (figure 6).43 Further HRCT features include the presence of bronchi within 1 cm of the costal pleura, the presence of bronchi abutting the mediastinal pleura and bronchial wall thickening. An objective score can be obtained by assessing each lobe separately for the degree of bronchial wall thickening and dilatation, mucous plugging, air trapping and consolidation.44
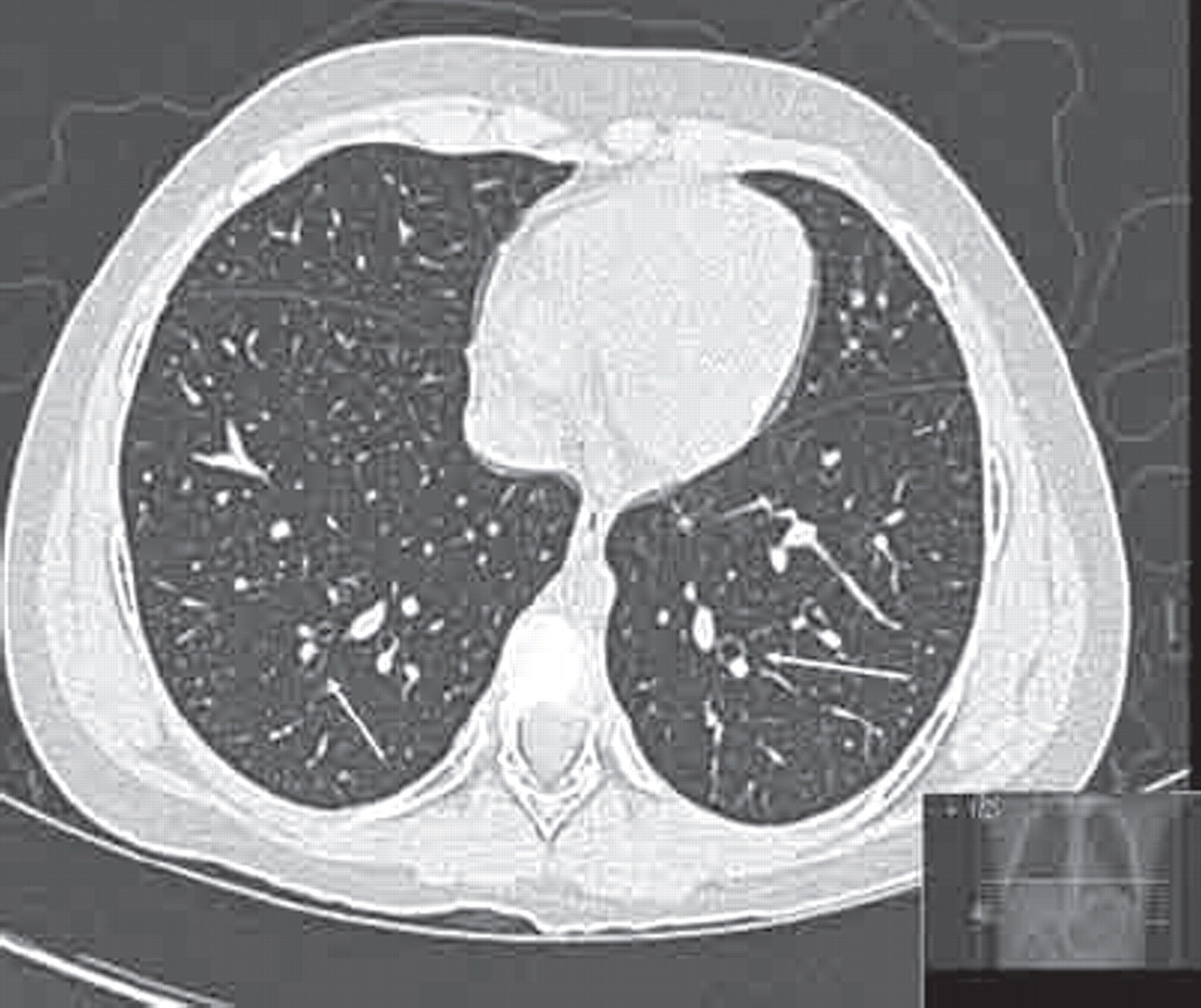
Dilated bronchi with smaller adjacent pulmonary vessel (“signet ring sign”) (arrows).
Li et al have suggested in their recent retrospective study of 136 children with bronchiectasis that the distribution of abnormalities on HRCT is not linked to aetiology.15 A lack of disease progression on follow-up HRCT scans at least 2 years apart has been demonstrated in a retrospective case notes review of 18 children with primary immunodeficiency.45 This is consistent with the observation that growth and spirometry often remain stable over time once treatment is instituted.46
Functional status ought to be evaluated using a combination of clinical assessment, radiographic features, spirometry and exercise testing.47
Investigating the aetiology
While investigating to confirm bronchiectasis with chest radiography and HRCT, the underlying cause should also be sought. The many aetiologies already described earlier can be investigated, often targeted in accordance with the accompanying history. Standard basic investigations are listed in table 1. It should be remembered that an underlying aetiology is often not found.
Basic investigations for the aetiology of bronchiectasis
A new classification
Heightened awareness of bronchiectasis and better availability of CT imaging has led to earlier identification and the need to question the concept of irreversible bronchiole dilatation.48 49 In their paper reviewing 93 children with non-cystic fibrosis bronchiectasis, Eastham et al propose subdividing chronic suppurative lung disease into three stages of bronchiectasis. Their definitions, although not yet universally accepted, are summarised below.11
Pre-bronchiectasis
“Chronic or recurrent bacterial endobronchial infection which may be associated with non-specific changes such as bronchial wall thickening on the HRCT scan. This condition may persist, resolve, or progress to “HRCT bronchiectasis”.” These changes are minimal and there is controversy whether in the absence of bronchial dilatation the label of “pre-bronchiectasis” can be used.
HRCT bronchiectasis
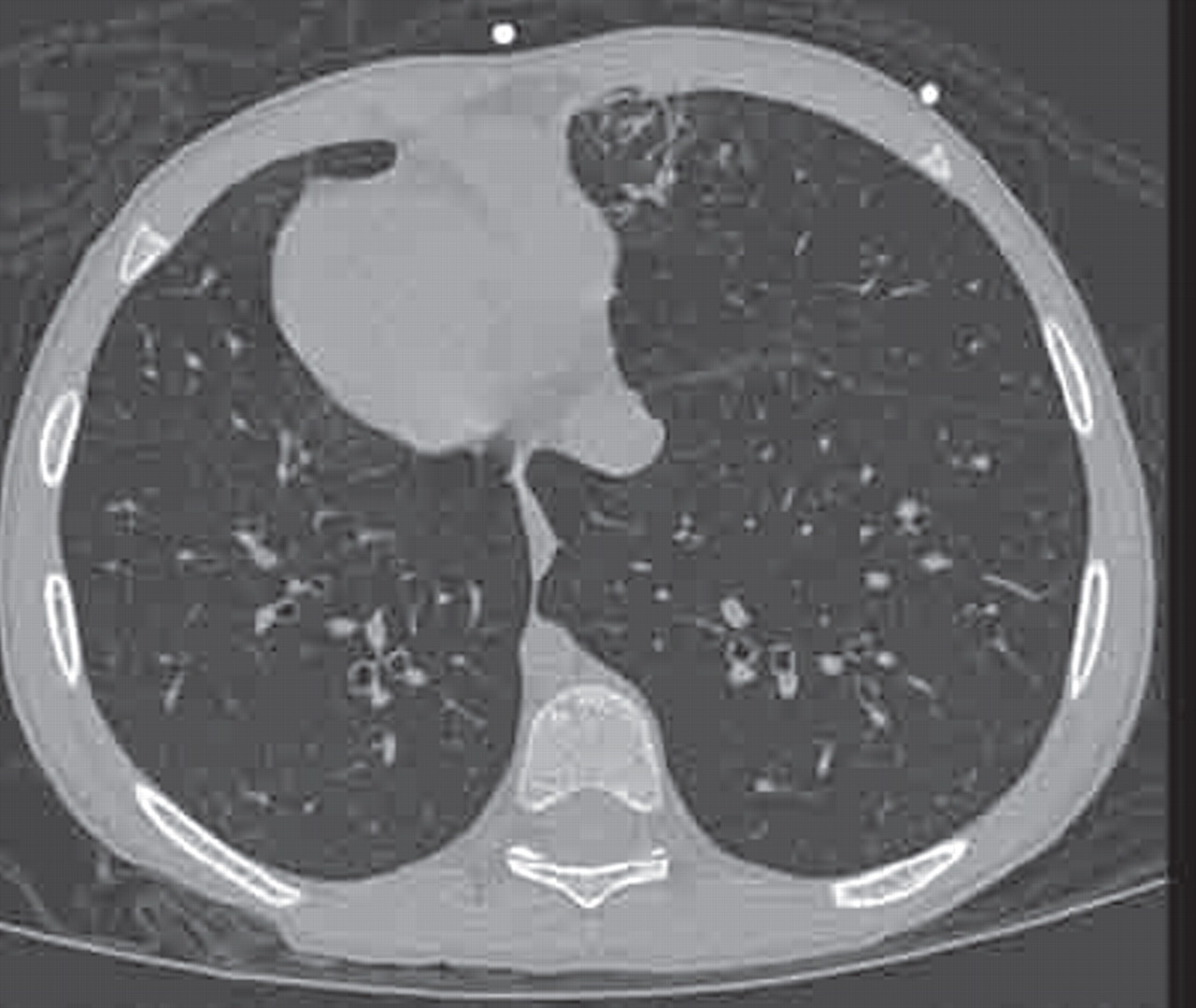
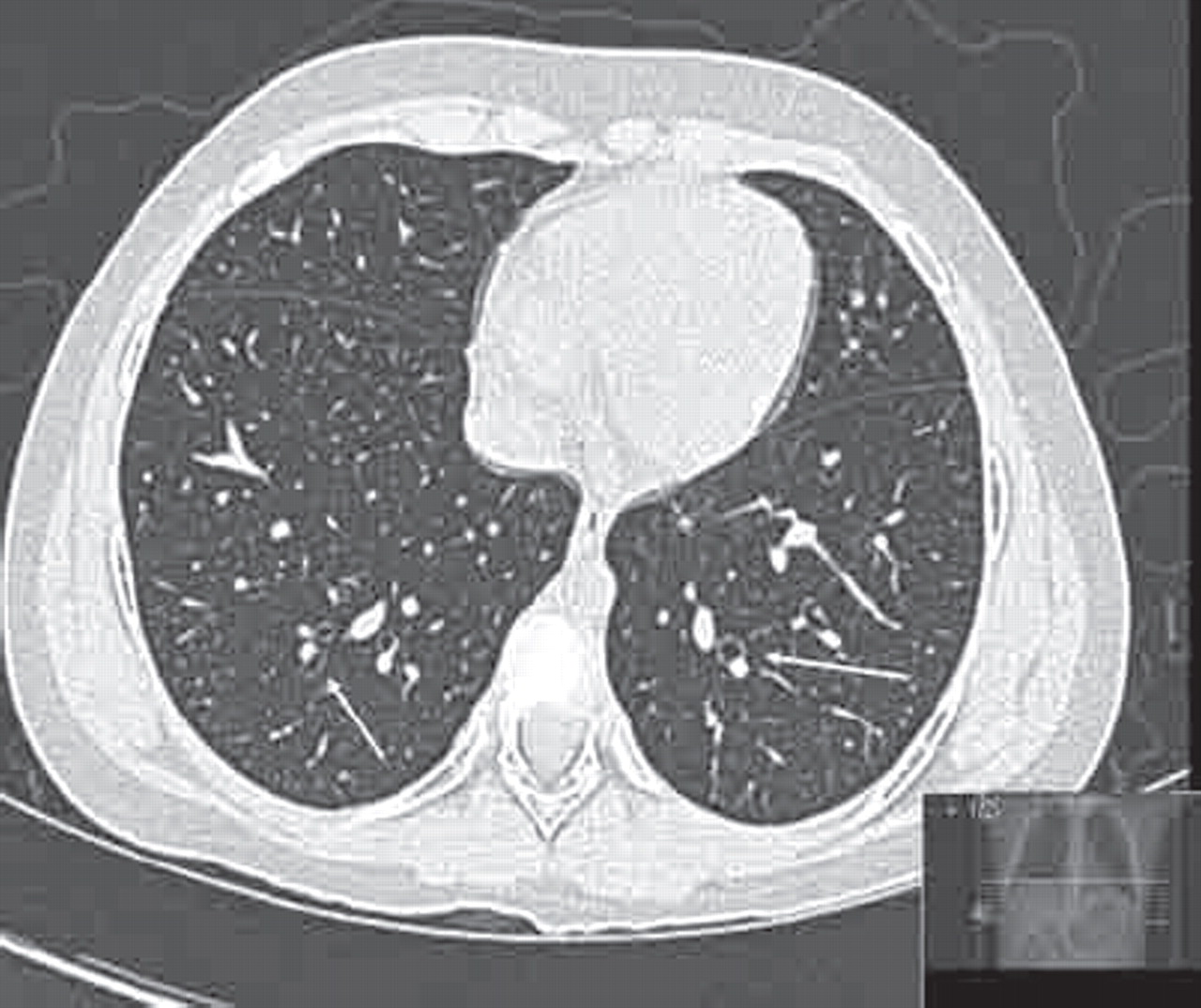
“The clinical features are associated with HRCT evidence of bronchial dilation.” An example of mild bronchial dilatation with no saccular or cylindrical changes is shown in figure 7. Eastham et al feel that this entity may “persist, progress to established bronchiectasis, return to a pre-bronchiectatic state or resolve entirely”. They suggest a repeat scan after a 2-year interval; if changes are still present they should be regarded as irreversible, especially in conjunction with recurrent respiratory infections and sputum production.

High-resolution computed tomography (HRCT) bronchiectasis showing thickened bronchioles with mild dilatation (arrows).
Established bronchiectasis
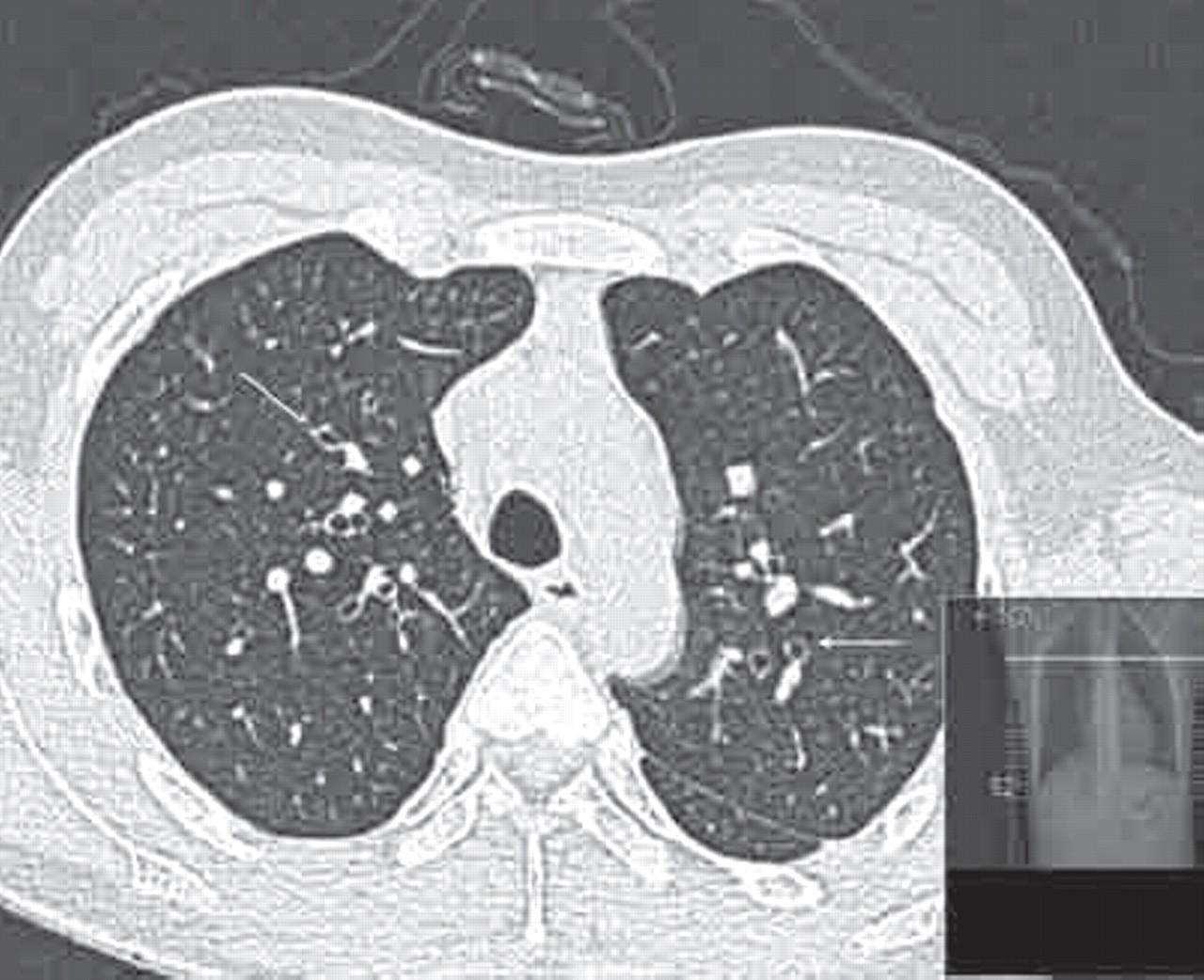
“The HRCT findings have not resolved after a significant time period.” Changes are then considered irreversible; an example of extensive established bronchiectasis is shown in figure 8.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
High-resolution computer tomography (HRCT) showing widespread established bronchiectasis.
Management
Long-term data characterising the natural history of the disease and high-quality studies into the effectiveness of treatments are scarce, so decisions are often based on expert opinion and extrapolation from other respiratory conditions, in particular cystic fibrosis. Results from adult studies are disappointing. In a recent cohort study of 76 patients, there was no significant effect of long-acting inhaled bronchodilators, inhaled or oral steroids, oxygen therapy, secretion clearance manoeuvres or antibiotics on the decline of forced expiratory volume in 1 s (FEV1).50
Vaccination
In September 2006 the 7-valent pneumococcal conjugate vaccine Prevenar was introduced into the routine childhood immunisation programme for all children from 2 months of age in the UK. Interestingly, while reports of invasive pneumococcal disease (IPD) due to any of the seven serotypes in Prevenar have seen a dramatic reduction, reports of IPD due to non-included serotypes are on the rise (www.hpa.org.uk). The effectiveness of pneumococcal vaccine as routine management in childhood bronchiectasis has been evaluated in a Cochrane review.51 Only one small non-randomised controlled trial in children was identified. It described a benefit in elimination of Streptococcus pneumoniae in the sputum but no clinical effect. The reviewers concluded that there was circumstantial evidence to support the use of routine 23-valent pneumococcal vaccination in children with bronchiectasis.
While the conjugated Haemophilus influenza vaccine used for primary immunisation in the current UK scheme offers protection against the encapsulated strain which poses the greatest risk for meningitis, respiratory infections are more commonly caused by non-encapsulated strains.
Despite the lack of evidence for or against routine annual influenza vaccination for children with bronchiectasis,52 this continues to be widely recommended based on a reduction in influenza related chest exacerbations seen in chronic obstructive pulmonary disease in adults, as half of the patients have co-existent bronchiectasis.53
Physiotherapy
Chest physiotherapy is now an established treatment for bronchiectasis, although research evidence for the value of physiotherapy is hard to find. Two small trials (n≤10 participants each) in adults with chronic obstructive pulmonary disease and bronchiectasis demonstrated a beneficial effect of postural drainage plus percussion54 and postural drainage plus forced expiratory techniques55 on sputum production and radioisotope clearance. The diversity of physiotherapy techniques and outcome measures make pooling of studies for the purpose of meta-analysis very difficult. Comparisons with placebo or no treatment failed to produce significant effects on pulmonary function.56
Improved sputum clearance can give patients who are established sputum producers an immediate feeling of clinical benefit. The theory behind physiotherapy is not only lung clearance but the hope that removal of sputum blocking the airways will allow less infection and hence less inflammation and prevent further damage. The importance of regular exercise as a form of physiotherapy cannot be over-emphasised.
Antibiotics
Contrary to the prevailing trend to reduce antibiotic prescription rates for respiratory tract infections in the general population,57 patients with bronchiectasis require regular microbiological surveillance and prompt treatment.
Frequent microbiological samples (sputum or cough swabs)should guide treatment and induced sputum or bronchoalveolar lavage may be indicated when samples cannot be obtained or remain negative in the face of recurrent exacerbations. A recent Cochrane review including 378 patients showed a significant beneficial effect of prolonged antibiotic therapy (between 4 weeks and 1 year) on response rates as reflected by reduced sputum volume in purulent bronchiectasis.58 It must be noted that only two out of the nine trials identified in the review included children and there was no difference between antibiotics and placebo with respect to reduction in exacerbation rates, lung function or mortality. The failure of this review to unequivocally support the use of antibiotics can be explained by the diversity of outcome measures in the included trials limiting meta-analysis. Furthermore, mortality can be expected to be extremely low in both groups, rendering it a poor discriminator.
In children, organisms most frequently isolated include Staphylococcus aureus, H influenzae and S pneumoniae,11 15 17 whereas Pseudomonas aeruginosa becomes more prevalent in adults59 which may be a reflection of increasing lung damage. A practice of zero tolerance to the presence of any respiratory pathogens should be adopted with an attempt at eradication with appropriate antibiotics; this may involve a combination of inhaled and oral antibiotics, often with the need for intravenous treatment.
Nebulised antibiotics, in particular for treatment of P aeruginosa, have been used for many years and are now easier to administer with the advent of more time efficient nebulisers. They have shown multiple benefits60,–,62 and exhibit a favourable risk benefit ratio, in particular as regards long-term aminoglycoside toxicity.
Thickened bronchial walls limit intraluminal bioavailability and antibiotics with high penetrance such as macrolides and quinilones are preferentially used. P aeruginosa, associated with accelerated lung function decline in cystic fibrosis, has a detrimental effect on prognosis in non-cystic fibrosis bronchiectasis as well.50 63 64
For recurrent infections prophylactic strategies can be employed. The appropriate choice of drug is informed by sputum culture results and azithromycin is most commonly used following successful trials in patients with cystic fibrosis.65,–,68
Macrolide antibiotics
The use of azithromycin is not only favoured because of its well-established direct antimicrobial effect on Gram positive cocci and atypical pathogens but also because macrolides appear to possess anti-inflammatory properties.69 70 Some bacteria intercommunicate through a mechanism called quorum sensing to create a biofilm which covers the airway epithelium and protects them from antimicrobials. Macrolides disrupt this process as well as decreasing the production of mucus and biosynthesis of pro-inflammatory cytokines.71
Four randomised controlled trials in cystic fibrosis have demonstrated an increase in FEV165,–,68 following treatment with macrolides. A recent study enrolled 34 children with steady-state bronchiectasis not due to cystic fibrosis or primary immunodeficiencies. The children were randomised to oral clarithromycin with routine management or routine management alone for 3 months. In the study group there was a significant reduction in neutrophil count and IL-8 in the bronchoalveolar lavage fluid at the end of the study period. No effect on FEV1 was detected.72 The long-term use of macrolides is of some concern because it can lead to acquisition of resistance in normal flora73; erythromycin-resistant S aureus and Haemophilus spp have been isolated in the sputum of cystic fibrosis patients following long-term administration of azithromycin.74
Anti-inflammatory drugs
Although a reduction in inflammation carries the potential of breaking the pathophysiological cycle (see figure 1), anti-inflammatory drugs have so far not been shown to be of significant benefit. There are currently no relevant trials evaluating the use of inhaled/oral corticosteroids, leukotriene receptor antagonists or non-steroidal anti-inflammatory drugs (NSAIDs) in paediatric bronchiectasis.
Immunoglobulin therapy
Immunoglobulin replacement therapy is used in children with established immunodeficiency, of either total immunoglobulins or functional deficiencies of antibody response. A high trough level of immunoglobulin is aimed at to stop further bronchiectasis occurring.
Bronchodilators
Small airway disease can be a component of bronchiectasis, but since a reduction in airway tone can potentially lead to increasing retention of secretions, its presence ought to be demonstrated by bronchodilator reversibility. Indeed, asthma can be present alongside bronchiectasis.
Short acting β-2 agonist therapy is the most frequently used treatment, with long-acting bronchodilators being prescribed more frequently. There are no randomised controlled trials to support this practice.75 76
Anticholinergic drugs are used effectively in obstructive airway disease for their effect on bronchoconstriction and bronchial secretions in adults. A Cochrane review identified 12 randomised controlled studies exploring the effect of anticholinergic therapy on acute exacerbations and stable bronchiectasis. None of them met the inclusion criteria, so currently no formal recommendations can be made.77
Mucolytics and hyperosmolar agents
Expectoration of infected mucus can be facilitated by changing the physiochemical properties of sputum. This approach is successfully used in cystic fibrosis. In non-cystic fibrosis bronchiectasis, there are insufficient data to make clear recommendations.78 79
Recombinant human DNase (rhDNase) breaks down the DNA that is released by neutrophils. In a double-blind placebo controlled trial involving adult patients, rhDNase administration resulted in no difference in FEV1 or forced vital capacity compared to placebo.80 In another randomised controlled trial in adults with stable state bronchiectasis, rhDNase was even reported to have a significant negative effect on FEV1 as well as more adverse effects including flu-like symptoms and higher exacerbation rate. Placebo treated patients used less antibiotics and steroids.81
Inhaled dry powder mannitol and hypertonic saline stimulate coughing and increase the amount of airway surface liquid, altering mucus rheology in a way that favours mucociliary clearance. There are no studies including children, but in adults dry powder mannitol was shown to improve airway clearance in central and intermediate, although not peripheral, lung regions.82 Cough transportability and short-term health status were also improved, although lung function remained unchanged.83 Hypertonic saline has not yet specifically been tested.
Surgery
There is no level 1 evidence for the use of surgery in children with bronchiectasis.84 Nevertheless, in symptomatic patients with localised disease (eg, following foreign body inhalation, bronchopneumonia or bronchial abnormalities) and insufficient benefit from maximal medical therapy, resection of the most significantly affected lobe is sometimes warranted to reduce the risk of overflow infection.
In a retrospective review of 54 children who underwent pulmonary resections, mostly in the form of lobectomy, complete resection of the bronchiectatic segment was achieved in 76% and 85% reported improvement or resolution of symptoms. Deterioration occurred in 9.4% and the mortality rate was 5.6%, mostly in patients with Kartagener's syndrome.85
A decision concerning surgical intervention requires careful deliberation in the framework of a multidisciplinary team involving respiratory physicians, cardiothoracic surgeons and radiologists. Although most centres routinely opt for thoracotomies, thoracoscopic surgery allowing earlier mobilisation and less post-operative pain has been found to be safe and effective in a recent case series including 19 patients aged 14 months to 22 years.86
Conclusion
Bronchiectasis in children is diagnosed more frequently than previously and in the early stages of bronchiole dilatation when mild it may be reversible, contrary to earlier notions. There is a paucity of quality evidence for most of the therapies available. The mainstay of treatment currently consists of aggressive use of antibiotics and physiotherapy (box 2). Most treatments have become established following extrapolation from strategies successfully used in cystic fibrosis. Large longitudinal studies are needed to characterise the natural history and progression of bronchiectasis and assess treatment effects on exacerbation rates, lung function, quality of life and mortality (box 3).
Box 2 Key points of management
High index of suspicion – chronic wet productive cough
Establish the diagnosis with a HRCT
Search for an underlying cause (eg, gastro-oesophageal reflux, immunodeficiency)
Treat underlying cause
Treat any exacerbation of respiratory symptoms (eg, increased cough, increased sputum) aggressively with appropriate antibiotics and augmented chest physiotherapy
Regularly monitor respiratory pathogens (eg, cough swabs, sputum) to guide treatment choice
Regularly monitor progress – lung function trends, plain chest x-ray, consideration of repeat HRCT, especially if there are early changes
HRCT, high-resolution computed tomography.
Box 3 Research pointers
Airway inflammatory cells need to be better characterised to help target treatment.
Randomised controlled trials are needed to assess utility of mucolytics, anti-inflammatory treatments and prophylactic antibiotics.
A universally agreed nomenclature for the distinction of different stages of bronchiectasis needs to be agreed.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.