Article Text

Abstract
Background Non-cystic fibrosis bronchiectasis is characterised by irreversibly dilated bronchi usually associated with chronic sputum production, bacterial colonisation of the lower respiratory tract, inflammation and frequent exacerbations. Irrespective of the underlying cause, this represents failure of the host defence to maintain sterility of the respiratory tract.
Objective To review the interactions and associations of non-cystic fibrosis bronchiectasis with the inate and adaptive immune systems with particular emphasis on known failure of local defences established deficiencies of the adaptive immune system. In addition we wished to explore potential subtle changes in the host defence which can lead to bacterial colonisation together with bacterial factors that aid colonisation of the lower respiratory tract and impair antibiotic response. This latter concept is considered with particular reference to Pseudomonas aeruginosa, which is often found in the airway secretions of patients with non-cystic fibrosis bronchiectasis and may act as a model for other organisms.
Methods An extensive literature review was undertaken to provide a comprehensive review of immunity and bacterial colonisation in non-cystic fibrosis bronchiectasis, with focus on in vitro studies examining bacterial factors which may facilitate colonisation together with potential implications for management.
Conclusion These themes provide a review of the current understanding of non-cystic fibrosis bronchiectasis together with areas for future research and potential therapeutic strategies.
- Bronchiectasis
- immunodeficiency
- inate immunity
- respiratory infection
- COPD mechanisms
- α1 antitrypsin deficiency
- bacterial infection
- COPD exacerbations
- COPD pathology
- cytokine biology
- emphysema
Statistics from Altmetric.com
- Bronchiectasis
- immunodeficiency
- inate immunity
- respiratory infection
- COPD mechanisms
- α1 antitrypsin deficiency
- bacterial infection
- COPD exacerbations
- COPD pathology
- cytokine biology
- emphysema
Introduction
Bronchiectasis is a pathological condition of lung damage characterised by inflamed dilated and thick-walled bronchi. It may be localised or diffuse, with the underlying cause identifiable in about 50% of cases.1 Clinical features of bronchiectasis include chronic production of sputum often mucopurulent or purulent in nature, persistent bacterial colonisation and recurrent lower respiratory tract infections. Symptoms include haemoptysis (especially during exacerbations) and breathlessness characterised by mild to moderate airflow obstruction, lethargy and reduced health status.2
Underlying conditions associated with bronchiectasis are summarised in table 1.
Causes of bronchiectasis
Whatever the cause, patients with bronchiectasis are often colonised with bacterial species, even in the stable state. This indicates failure of the usual host defences that have evolved to maintain sterility of the respiratory tract. The purpose of the current article is to provide a review of bacterial and host factors in more classical idiopathic bronchiectasis in order to identify pathophysiological features that may explain the propensity for bacterial colonisation.3 In particular, we wish to explore more subtle changes in host defence as well as bacterial factors that may facilitate colonisation and/or impair antibiotic response. These data may provide a more rational approach for the future management of the disease and provide insight into the problems of bacterial colonisation of the lower respiratory tract in general.
Immune system and its interactions/associations with bronchiectasis
The concept of the ‘vicious cycle’ hypothesis of bronchiectasis implicates failure of host defence leading to a host-mediated chronic inflammatory response causing new or further impairment of mucociliary clearance and host defences, thereby amplifying the problem.4 The interplay between organisms and host defence represents a frustrated attempt at clearance, leading to excessive inflammation (in some), further impairment of host defences, and establishing and maintaining a vicious cycle.
Of particular interest in bronchiectasis are the bacteria which commonly colonise the lung and especially Pseudomonas aeruginosa (P aeruginosa), which interacts with the host, and has immunomodulatory properties that facilitate its survival. These features of the immune system in bronchiectasis will be particularly explored below.
The overall interaction between lung host defences and bacteria is summarised in figure 1.
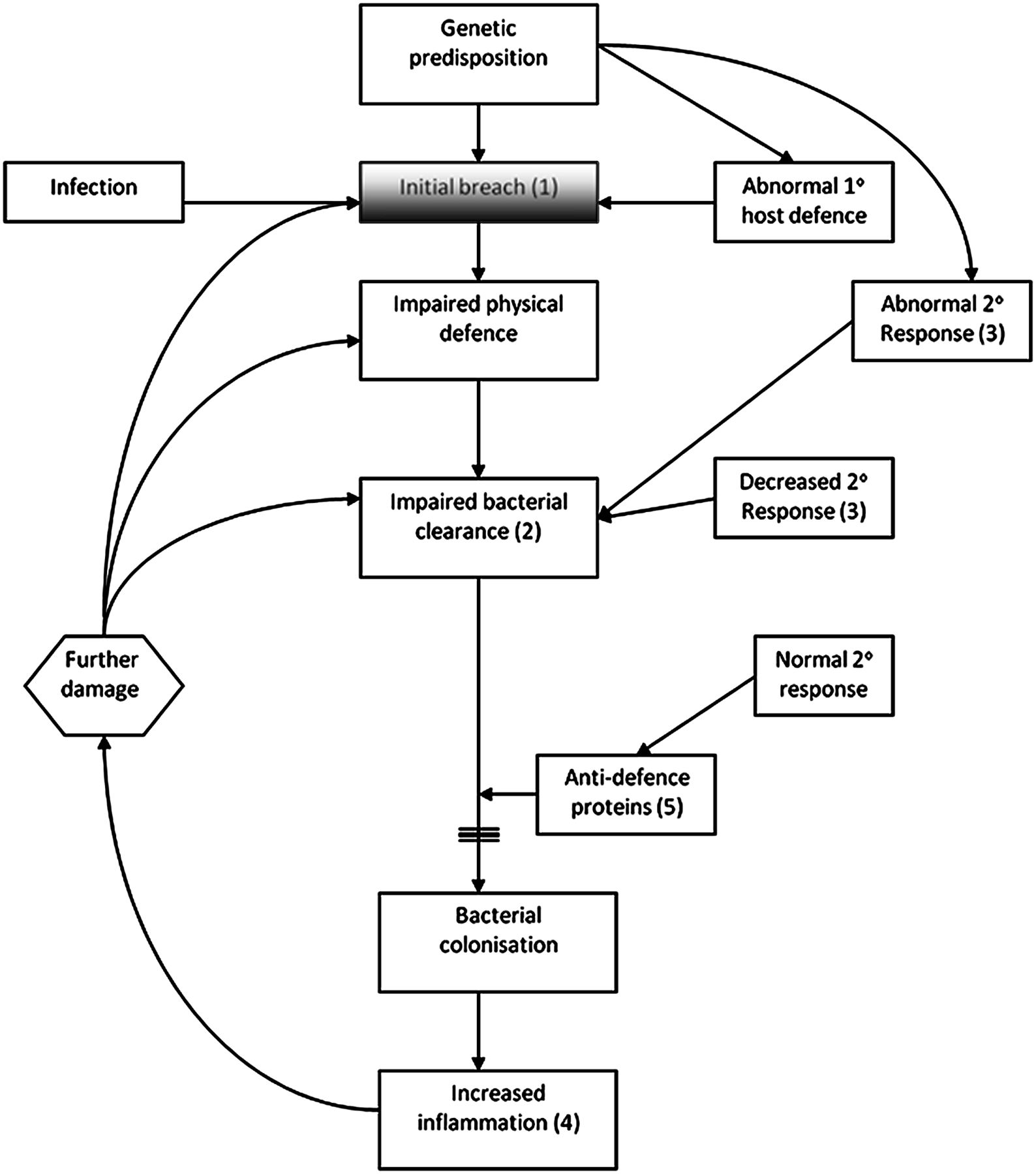
Interactions between lung host defence and bacteria. This diagram demonstrates the breach in the lung defence precipitated either by a genetic predisposition, infection or abnormal primary host immune defence (innate immune system). The initial breach (1) leads to further impairment of the host defence with impaired bacterial clearance (2). Impaired bacterial clearance can also be initiated by a decreased or abnormal secondary host immune response (adaptive immune system) (3) with failure of the system to respond to an insult with a ‘learned’ response. The normal secondary response can also be impaired by further damage. This impairment of bacterial clearance leads to a state of bacterial colonisation and ongoing and increased inflammation as a result of continuing release of anti-defence proteins. As previously described by Cole et al 4 the vicious cycle is set in motion and a state of continuing infection and inflammation (4) exists with progressive lung damage. A normal and intact secondary response (5) ought to prevent bacterial colonisation and further damage.
The innate immune system of the lung
The innate immune system is the immediate, non-specific and first line of defence against invading organisms. The mucociliary escalator is lined by ciliated and mucus-producing cells. Particles >2–3 μm impact on the mucus covering epithelial cells in the upper airways and bronchi and are removed by a combination of normal mucociliary clearance and cough.5 ,6 Airway secretions also contain several anti-infective proteins and polypeptides, including the bacteriostatic proteins lysozyme and lactoferrin. Secretary leucoprotease inhibitor (SLPI), produced by the mucous glands, macrophages and epithelial cells, has antifungal, antiviral and antibacterial properties.7 The role of antimicrobial proteins and peptides is discussed later together with neutrophils and their role in bronchiectasis. Lactoferrrin, SLPI, lysozyme and defensins are (among others), derived from recruited neutrophils in addition to submucosal glands (for lactoferrin).7
Secretary IgA is the main antibody isotype in mucosal secretions in the respiratory tract that contributes to the initial defence mechanisms, while IgG predominates in the plasma.8 Most lung IgA is produced locally within the pulmonary tissues by subepithelial plasma cells. Postmortem studies of lung tissue indicate an increased number of IgA-positive cells in bronchitic and bronchiectatic lungs compared with controls.9
The proportion of the IgA subclasses is different in lung secretions and blood. Most of the blood IgA is of the IgA1 subclass with 10–20% being IgA2, whereas lung secretions have around 30% IgA2. Stability of IgA2 is maintained better than IgA1 in the presence of infection because of resistance to IgA proteases produced by many pathogenic bacteria.9 There is increased production of IgA2 in the presence of infection indicated by an increase in the proportion of IgA2 with increasing sputum purulence.10 IgA in airway secretions binds to lectin-like bacterial adhesins blocking epithelial colonisation and operating as a generic immune barrier. In addition, IgA promotes phagocytosis by binding to Fc receptors expressed on the surface of eosinophils, neutrophils and monocytes.9 IgA enables delivery of antigens across the epithelium into luminal secretions, thereby removing locally formed immune complexes and limiting access to the systemic circulation. Mucosal IgA production is maintained by factors such as interleukin (IL)-2, IL-5, IL-6 and IL-10 generated by airway epithelial cells.9
If these primary clearance mechanisms are overwhelmed by a high bacterial load, a secondary defence system is activated. Animal experiments clearly demonstrate this relationship between bacterial load and activation of the secondary host response in that a low bacterial load leads to bacterial clearance, but when the bacterial load is greater than 106 colony-forming units/ml (cfu/ml) there is a neutrophilic host inflammatory response.11
Failure of the local host defences results in microbial colonisation, establishing the ‘vicious cycle’ described above. Airway epithelium responds by releasing antimicrobial peptides and inflammatory cytokines into the airways. In human airway epithelial cells the main classes of antimicrobial peptides produced are the β-defensins and the cathelicidins (IL-37 in humans). Chemokines and cytokines such as IL-8 are released into the submucosa12 and initiate the inflammatory reaction, leading to recruitment of phagocytes (neutrophils, monocytes and macrophages), dendritic cells and lymphocytes, which contribute to the adaptive response.13 The airways become inflamed leading to leakage of serum proteins including circulating antibodies and complement, therefore increasing local antibacterial potential.
Role of neutrophils and neutrophilic inflammation in bronchiectasis
Neutrophil-dominant inflammation is a prominent feature in bronchiectasis. Neutrophils aggregate rapidly at the site of infection with three key actions. Namely, phagocytosis, the release of the contents of pre formed granules, and production of reactive oxygen species (ROS), all of which are directed at bacterial killing.14 ,15
The internalisation of particles or bacteria and the formation of a phagosome are facilitated by opsonisation, which is when IgG antibodies bind to organisms by the Fab portion, with the Fc portion then binding to the phagocyte via specific receptors. Complement system proteins are activated either through Ig binding (classical pathway) or independently (alternative pathway), emphasising the importance of immunoglobulin and complement deficiency in the pathophysiology of bronchiectasis. Low or deficient serum Mannose-binding lectin activating protein of the lectin pathway of the complement system has also been associated with bronchiectasis.16 ,17
Normally the neutrophil moves to the site of phagocytosis in response to chemotactic stimuli. In bronchiectasis, leukotriene B4 and IL-8 in addition to other chemotactic factors have been implicated, with IL-8 playing a more crucial role during acute exacerbations.18
Additionally, the granule proteins, formed during neutrophil development, and released on activation, have a vital role in neutrophil response.19 The three subsets of granules contain a variety of enzymes and host defence proteins with receptors, signalling proteins and adhesion molecules that become expressed on their membrane following activation. Through a hierarchical process of degranulation, azurophilic granules (primary granules) release (among others) myeloperoxidase, bactericidal/permeability-increasing protein, defensins, neutrophil elastase, proteinase 3 and cathepsin G. Specific granules (secondary granules) release other key antibacterial proteins such as lactoferrin and cathelicidin and tertiary (gelatinase) granules contain, and release, gelatinase acetyltransferase and lysozyme. The excessive degranulation and activity of cytolytic enzymes such as myeloperoxidase and elastase have been implicated most in host tissue damage.20
The respiratory burst results in the release of ROS to facilitate bacterial killing. The release of uncontrolled ROS by neutrophils may however also lead to damage to surrounding tissues, amplifying the lung disease process.21 It had previously been thought that ROS exert a direct toxic effect on ingested pathogens. However Reeves et al 22 suggested that the respiratory burst sets in motion events which result in creating a favourable environment for proteases such as elastase to digest the contents within the phagocytic vacuole. By optimising conditions via ion flux and pH change, the enzymes released from the cytoplasmic granules become more active, facilitating the destruction of ingested pathogens.
The respiratory burst is based upon the assembly and activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.21 The genetic defect of chronic granulomatous disease emphasises the importance of this pathway. It is caused by a defective gene for one of the subunits of NADPH oxidase and leads to an inability to produce ROS, resulting in recurrent life-threatening bacterial infections and granuloma formation.23 Patients with this condition frequently develop a variety of inflammatory complications consistent with exaggerated inflammatory responses including bronchiectasis.24
In 2006, King et al 25 demonstrated that phagocytosis was normal in patients with bronchiectasis compared with controls, but that oxidative burst was lower. It was suggested this was either due to a suppressive effect of bacterial products on neutrophil function or an intrinsic defect of neutrophil oxidative burst. However, earlier work by Pasteur et al 1 failed to demonstrate any significant difference in neutrophil respiratory burst. It remains possible that this represents an important abnormality but that its frequency depends on patient referral patterns. The basic physiological defect has yet to be identified.
Toll-like receptors
Toll-like receptors (TLRs) are a family of cell surface immune-modulatory proteins playing a crucial role in both the innate and adaptive immune response. They induce an immune response to a number of pathogenic factors with 10 human TLRs identified to date. These receptors play a vital role in recognising pathogens and their products, and thereafter activating the immune response.
TLRs recognise bacterial structures called pathogen-associated molecular patterns (PAMPs)26 and TLR2, TLR4, TLR5, TLR6 and TLR9 have been specifically implicated in the recognition of bacterial components. TLR2 is expressed in human alveolar epithelial type II cells and alveolar macrophages,27 and recognises peptidoglycan and lipoprotein, playing a role in mediating the inflammatory response to Gram-positive and mycobacterial products.28 TLR4 is outer membrane associated and responsible for the recognition of lipopolysaccharide (LPS). TLR9 is found on the surface of endosomes and responds to nucleic-acid-based PAMPS of bacteria and viruses, whereas TLR5 is an outer membrane receptor for bacterial flagellin.29 ,30 Table 2 summarises the TLRs and the pathogens they respond to via PAMPs.
Toll-like receptors (TLRs) and their pathogens31
The interaction between the TLRs and PAMPs leads to activation of the innate host defence, especially the release of proinflammatory cytokines as demonstrated by lack of response in TLR knockout mice.32 In health, the interaction of PAMPs and TLRs is necessary for an appropriate host response to the specific pathogen.
This process has been clearly demonstrated for P aeruginosa and its interaction with TLRs, which is of particular importance in bronchiectasis given the morbidity associated with colonisation by this organism.33 P aeruginosa is an opportunistic Gram-negative organism which is a recognised colonising organism in both idiopathic bronchiectasis and cystic fibrosis (CF), often in more severe bronchiectasis.34 ,35 Chronic colonisation with P aeruginosa has also been identified as an independent risk factor for an accelerated rate of decline in lung function.33
P aeruginosa LPS is recognised by TLR432 and induces lung inflammation in vivo.36 In addition, TLR5 recognises flagellin expressed by Pseudomonas, with signalling assistance from TLR2,37 leading to proinflammatory cytokine and chemokine production resulting in accumulation of neutrophils in the air spaces.36
Skerrett et al 36 demonstrated (in mice) that TLR4 and flagellin-induced signals mediated most of the immune response to Pseudomonas and that TLR2 had a counter-regulatory role.They demonstrated a diminished tumour necrosis factor (TNF)-α response to P aeruginosa in bone marrow cells lacking TLR4 alone or with TLR2. Their studies have supported both specific and complimentary roles for TLR2, TLR4 and TLR5 in the recognition of P aeruginosa and shown that signalling via these receptors accounts for most of the early cytokine and inflammatory response to infection.
Moreover, TLR5 surface expression has been shown to be upregulated on respiratory epithelial cells following exposure to P aeruginosa flagella.37 TLR5 is stored intracellularly in neutrophils but activation of TLR1 and TLR2 mobilises the receptors to the cell surface. It has been proposed that TLR5 plays a vital role in the interactions between P aeruginosa and neutrophils in CF. Koller et al 38 confirmed this by demonstrating that activation of TLR5 by flagellin enhanced the production of IL-8, phagocytosis and respiratory burst by neutrophils. TLR4-deficient mouse models show a delay in clearance of Haemophilus influenza, which also colonises patients with bronchiectasis, following intrapulmonary administration with a diminished inflammatory response, suggesting a crucial role for this receptor in mediating an infective immune response to H influenzae. 39 The role of oligosaccharides from non-typeable H influenzae remains to be determined.
The role of TLRs in airway neutrophilic inflammation has been assessed in patients with asthma and bronchiectasis. In patients with neutrophilic asthma there was increased expression of TLR2 and TLR4 compared with controls and patients with eosinophilic and paucigranulocytic asthma. In patients with bronchiectasis, increased TLR2 mRNA was found in induced sputum but there was no difference in the expression of TLR4. It was suggested that the data may indicate differential expression of TLRs in bronchiectasis.40 Whether this represents cause or effect remains to be determined.
Animal models of emphysema have also demonstrated increased expression of TLR2 and TLR4 in airway macrophages.41 It was proposed that this increased expression may enhance the immediate inflammatory response following inoculation with Streptococcus pneumonia. Whether this is relevant to bronchiectasis also remains to be determined, but again S pneumoniae is often identified as a colonising organism.
Acquired immune system
The adaptive immune response is required to protect the lung against organisms that evade phagocytosis by macrophages leading to local replication.42 Marked defects of the adaptive immune response are adequately described elsewhere and thus only mentioned here briefly.
Primary antibody deficiency syndromes (PADS), all of which have been associated with bronchiectasis, involve partial or complete absence of one or more immunoglobulin class or subclass. In addition, there may be more subtle defects of specific responsive antibody formation,43 as summarised in table 3.
Primary antibody deficiency syndromes
The effect of common variable immune deficiency (CVID) is an inability to mount an appropriate and competent antibody response to pathogen epitopes. Kainulainen et al 44 found that bronchiectasis was present in 16 of 22 (73%) patients with either CVID or x-linked agammaglobulinaemia, with pathological progression in five patients over 3 years despite adequate immunoglobulin replacement therapy. Quinti et al 45 also demonstrated similar findings in 2007.
Evidence from patient series demonstrate overall increased survival in patients treated with immunoglobulin replacement. A number of patient series have also demonstrated reductions in acute and chronic infections in treated patients with PADs.46 ,47
The significance of IgG subclass deficiencies in bronchiectasis
IgG has four subclasses, each with different antibody responses. Low levels of isolated IgG subclasses have been identified, with undetectable levels of serum IgG4 present in up to 10% of healthy adults.48
Hill et al concluded in 1998 that IgG subclass deficiency was comparatively rare in patients with bronchiectasis compared with age-matched healthy controls.49 Similarly, King et al 25 showed no major abnormalities in testing of immunoglobulin levels in their cohort. Stead et al 50 also concluded that isolated antibody subclass deficiency was an uncommon cause or association with bronchiectasis in patients beyond the fourth decade.
More recently, Pasteur et al 1 found humoural immunodeficiency in 12 of 150 patients (8%), six of whom had isolated IgG deficiency. One patient was classified as having severe IgG2 deficiency with evidence of defective antibody production, and three had borderline low levels with evidence of defective antibody production alone. A further six patients had normal immunoglobulin levels but also evidence of isolated defective antibody production in response to antigen. The authors concluded that the relevance of isolated IgG subclass deficiency was not clear and that in addition to immunoglobulin and IgG subclass measurements, the need for functional studies was critical to investigate humoural immunity.
Similarly, Vendrell et al 51 supported this finding in patients with idiopathic bronchiectasis and normal IgG levels and concluded antibody deficiency with normal IgG levels may be associated with bronchiectasis. They advised evaluation of antibody response to both H influenzae (capsular antigen) and pneumococcal (protein antigen) vaccines.
The role of immunoglobulin therapy is clearest in patients with CVID or agammaglobulinaemia aiming to reduce acute and chronic infections and their sequelae. However, immunoglobulin replacement in patients with specific antibody deficiency or subclass deficiency is less clear. Immunoglobulin replacement is less likely to be useful in partial subclass deficiencies or IgA deficiency without associated defects52 and indeed may be detrimental. Patients may become positive for IgG anti IgA antibodies, after exposure to IgA in blood products, and develop a predisposition for anaphylactoid reactions to intravenous immunoglobulin.53
Blocking antibodies
Bactericidal blocking activity was described in the serum of patients with chronic Gram-negative infections by Waisbren and Brown in 1966, who suggested that the blocking factor may be an antibody.54 Subsequent work in the 1970s demonstrated the bacterial blocking activity in chronic P aeruginosa infection in CF was IgG mediated.55 Hornick and Fick56 explored this concept of ‘blocking antibodies’ further and demonstrated that the IgG subclass response to P aeruginosa LPS was largely restricted to subclass 2. Further, they identified IgG2-containing immune complexes which inhibited bacterial phagocytosis by pulmonary macrophages and neutrophils. They also demonstrated an inhibitory effect of IgG2 immune complexes in the sera of patients with CF and chronic P aeruginosa colonisation. The authors concluded that sera from these patients functioned poorly as an opsonin and that IgG2 antibodies exert an antiphagocytic effect both by direct inhibition and the formation of immune complexes.56 This concept is summarised in figure 2.

IgG2 and IgG2 containing immune complexes in the serum of patients with cystic fibrosis colonised with Pseudomonas aeruginosa. IgG2 has blocking activity by direct inhibition and the formation of IgG2-containing immune complexes in the serum of patients with cystic fibrosis and chronic P aeruginosa colonisation. Patients with cystic fibrosis and chronic P aeruginosa colonisation were found to have significantly elevated levels of IgG2. Phagocytosis by pulmonary macrophages is inhibited in the presence of serum from these patients while IgG2-containing immune complexes inhibit phagocytosis by pulmonary macrophages and neutrophils. It was concluded that sera from these patients does not function well as an opsonin. Patients with cystic fibrosis and chronic P aeruginosa colonisation also have elevated levels of these immune complexes. Removal of the immune complexes leads to a simultaneous fall in the levels of IgG2 and increased bacterial uptake by neutrophils and pulmonary macrophages. It is hypothesised that this may be due to competition by these immune complexes with opsonised bacteria for the Fc receptor sites on phagocytic cells. IC, immune complex.
Later work by Thomassen and Demko57 demonstrated that patients with CF, in poor clinical condition, were more likely to have Pseudomonas strains that were resistant to serum bactericidal activity in autologous serum than those in a better clinical state. These strains were however sensitive to normal healthy serum, suggesting the presence of blocking activity in the serum. It was hypothesised that the development of this resistance was a contributory factor in the failure of the host to control infection leading to a poorer clinical condition.57
More recently, the concept of blocking antibodies has been explored in detail in Salmonella infection in patients who are HIV positive. There is a high mortality associated with non-typhoidal strains of Salmonella in this patient group.58 The data demonstrated a lack of bactericidal activity as a result of dysregulated humoural immunity in the sera, with excess IgG directed against S typhimurium LPS. By extracting LPS IgG from inhibitory HIV-infected serum and adding this to HIV-uninfected serum, the anti-LPS IgG was confirmed as the inhibitor of S typhimurium killing. Further studies by the same authors demonstrated that this inhibition could be overcome by adding IgG from HIV-uninfected sera to inhibitory HIV-infected sera in appropriate proportions. These data clearly demonstrated the importance of competing blocking and killing antibodies in patients infected with Salmonella, although the subclass was not identified.58 Whether this plays a role in bronchiectasis remains to be investigated. However, of importance, antibodies to polysaccharides are usually of the IgG2 subclass, providing a potential link between the role of IgG2 in CF and the clinical effect of endotoxin-specific antibodies in Salmonella infection. Clearly further studies in both CF and non-CF bronchiectasis are indicated. In particular, the role of bactericidal blocking activity directed against P aeruginosa in these patients may provide some explanation for the propensity to chronic colonisation.
Quorum sensing and biofilm development
A number of specific bacterial factors enhance the ability of the pathogen to evade host defences. These include adherence, the ability to impair ciliary clearance and damage the epithelial surface (which enhances both), and avoidance of phagocytosis.36 Therefore, the balance between the innate and adaptive immune response and bacterial factors that avoid or subvert this response will result in either reduced bacterial clearance or facilitate proliferation leading to colonisation.
An area of increasing interest and research in bacterial behaviour is quorum sensing. This complex cell-to-cell signalling mechanism of bacterial populations modulates their behaviour and ability to survive, colonise and potentially invade the host. Quorum sensing is defined as the capacity to detect extracellular, small-molecule signals and to alter gene expression in response to bacterial population densities that reach a threshold concentration. These signalling pathways modulate various functions of the bacteria, including their metabolism, expression of virulence factors and biofilm formation.59
Many common bacteria contain quorum-sensing genes. Much of the research to date has specifically examined the quorum-sensing system employed by P aeruginosa. It is difficult to eradicate and therefore its behaviours mediated by quorum-sensing systems and potential interactions with the host defence are likely to be of major importance.
The quorum sensing of Gram-negative bacteria is predominantly via the well characterised N-acyl homoserine lactone (AHL) signalling system.60 ,61 The AHL-based circuits are encoded by two signalling systems operating with specific signalling molecules, termed the Las and Rhl system. The two systems operate with specific signal molecules: 3-oxo C12-HSL for the lasR-encoded receptor and C4-HSL for the rhlR-encoded receptor.60 ,61 These systems also interact with host defences. Telford et al 62 demonstrated that the Pseudomonas quorum-sensing signal molecule N-(3 oxododecanoyl)-L-homoserine lactone (3-oxo-C12-HSL) had immunomodulatory activity in addition to its recognised role in regulating virulence. Ritchie et al 63 showed direct effects on T cells and a role of this molecule in the initial stages of T-cell activation and Smith et al 64 demonstrated (in mice) that the quorum-sensing systems of Pseudomonas could induce inflammatory mediators in vivo via T cells, promoting the production of the inflammatory cytokine γ interferon.
In addition, 3-oxo-c12-HSL induces apoptosis in the neutrophils and macrophages. Tateda et al 65 proposed a mechanism of chronic Pseudomonas infection in the lungs involving quorum sensing whereby the formation of biofilms created a favourable environment for Pseudomonas survival. They suggested that 3-oxo-C12-HSL induced the production of neutrophil chemotactic factors (IL-8 in humans and macrophage inflammatory protein-2 in mouse models). This sets in motion a chain of events leading to tissue destruction and accelerated neutrophil apoptosis, all of which contribute to the persistence of Pseudomonas in the lungs. In the presence of 3-oxo-C12-HSL neutrophil apoptosis is accelerated, therefore reducing bacterial killing capacity by these cells. Incubating neutrophils with 3-oxo-C12-HSL was shown to increase markers of apoptosis. It was proposed that apoptotic neutrophils also provide nutrients for the bacteria, facilitating their proliferation. This concept is outlined in figure 3. It is not known whether these Pseudomonas products are active in the airways of colonised patients with bronchiectasis and further studies are clearly needed to establish whether this process is important in establishing and maintaining colonisation of P aeruginosa.

Proposed interactions of quorum sensing signal molecule N-(3 oxododecanoyl)-L-homoserine lactone (3-Oxo-C12-HSL) and the host defence. 3-Oxo-C12-HSL (1) is proposed to have immunomodulatory activities via T cells and promotes the production of inflammatory cytokines (γ interferon) and neutrophil chemotactic factors (interleukin-8 (IL8)/macrophage inflammatory protein-2 (MIP2)) (2) in addition to its recognised role in quorum sensing. Specifically it has been suggested that the production of these neutrophil chemotactic factors leads to accelerated neutrophil apoptosis (3). This, in turn, leads to persistence of pseudomonas by reduced killing by neutrophils due to early cell death, and it is proposed, the products of apoptosis provide nutrients for the bacteria, allowing its proliferation (4) and biofilm formation (5), protecting the bacteria further from phagocytes and antibiotics.65
One of the other important features of P aeruginosa is its ability to form biofilms. These are defined as a sessile community characterised by cells that are irreversibly attached to a surface or interface, or to each other, and embedded in a matrix of extracellular polymeric substance exhibiting an altered phenotype with respect to growth rate and gene transcription compared with planktonic bacteria.66 Biofilms protect the bacterial population against host defence and increase the tolerance to antimicrobials. This is emphasised by their ability to form in the lungs of chronically ill patients, on catheters and on prosthetic valves, often requiring prolonged periods of antibiotic therapy to eradicate the organism.60 ,67 Links between biofilm formation and quorum sensing have been well established,60 ,67 although there are a number of other physical, biological and chemical factors involved in biofilm development. For example, when biofilms form on biomedical devices the process begins with the coating of the device by proteins and glycoproteins, which may act as binding ligands.68 Furthermore, in vivo, manipulation of the medium in which P aeruginosa is grown has been shown to influence the motility of bacterial populations and therefore the ability to form biofilms or the type of biofilm formed. P aeruginosa has been shown to form mushroom-like structures in flow cell reactors69 However, in environments deplete of iron, flat biofilms are formed with wild-type P aeruginosa, which had previously grown in the classical mushroom form in an iron replete medium70 The primary source of carbon as a nutrient also influences the structure of the biofilm, which differs from those in which glucose is the main source rather than citrate. This is thought to be due to differences in the motility of the bacterial population influenced by the primary carbon source.71 By manipulating the quorum sensing of P aeruginosa populations and thereby creating las I mutants, the type and shape of biofilm formed is also influenced, with mutants forming flat biofilms compared with wild type.72 Subsequent work has suggested that the formation of biofilm, and its structure, is not solely dependent on quorum sensing but also on the medium in which the biofilm is cultivated.73 Biofilm formation therefore represents a highly complex and multifactorial process, which is summarised in figure 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
P aeruginosa biofilm life cycle. Stage 1: reversible attachment—individual bacterial cells populate the surface. Stage 2: irreversible attachment—extracellular polymeric substance is produced and attachment becomes irreversible. Stages 3–4: maturation phase—biofilm architecture develops and matures. Stage 5: dispersion—single cells are released from the biofilm and perpetuate the cycle. (Image reproduced with the permission of Peg Dirckx, Center for Biofilm Engineering, Montana State University, Bozeman, Montana, USA. ©MSU-CBE, P, Dirckx.)
The components of the adaptive immune system and how they interact with biofilms and quorum sensing signalling molecules is, at least, partly understood. In 2005 Walker et al 74 demonstrated enhancement of biofilm development by neutrophils. However, the exact role of quorum sensing in biofilm formation in idiopathic bronchiectasis is less clear than in CF.75 It should be noted that the P aeruginosa species of CF is usually of the mucoid phenotype. Whether the same processes are active in non-CF bronchiectasis, for which most of the P aeruginosa colonisation is non mucoid, remains to be determined. However, because P aeruginosa is a common colonising organism in both conditions and particularly in view of the difficulty in achieving microbial sterilisation in both, the data suggest that biofilm formation may also provide a protective bacterial mechanism in non-CF bronchiectasis.
Overall it is likely that quorum sensing and biofilm formation play a role in the ability of bacteria, and in particular P aeruginosa, to colonise the host but also contribute to its ability to persist in the lungs and subvert the host immune defence. The net result is excessive but ineffectual adaptive immunity which in itself may lead to perpetuation of bacterial colonisation, and continued neutrophil proteolytic and oxidative damage; that is the vicious cycle. Understanding these processes will be essential to the development of future effective therapeutic strategies, targeting their role in maintaining the vicious cycle.
Conclusion
In conclusion, it is clear that all aspects of the immune system and their perturbation by bacteria and their products are complex and central to the development, clinical presentation and management of bronchiectasis. Growing fields of interest and research include the roles of TLRs, blocking antibodies, quorum sensing and biofilms. Much of our knowledge so far has been obtained from animal models and infections outside of the lung, and the time has come to investigate the role of these factors in patients with bronchiectasis. The information obtained will be central to the future management of patients.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.