Article Text

Abstract
Background: Rhinoviruses (RVs) are the most frequent precipitants of the common cold and asthma exacerbations, but little is known about the immune response to these viruses and its potential implications in the pathogenesis of asthma.
Methods: Peripheral blood mononuclear cells (PBMC) from patients with atopic asthma and normal subjects were exposed to live or inactivated RV preparations. Levels of interferon (IFN)γ and interleukins IL-12, IL-10, IL-4, IL-5 and IL-13 were evaluated in the culture supernatants with specific immunoassays.
Results: Exposure of PBMC to RVs induced the production of IFNγ, IL-12, IL-10, and IL-13. Cells from asthmatic subjects produced significantly lower levels of IFNγ and IL-12 and higher levels of IL-10 than normal subjects. IL-4 was induced only in the asthmatic group, while the IFNγ/IL-4 ratio was more than three times lower in the asthmatic group.
Conclusions: This evidence suggests that the immune response to RVs is not uniquely of a type 1 phenotype, as previously suggested. The type 1 response is defective in atopic asthmatic individuals, with a shift towards a type 2 phenotype in a way similar, but not identical, to their aberrant response to allergens. A defective type 1 immune response to RVs may be implicated in the pathogenesis of virus induced exacerbations of asthma.
- rhinovirus
- asthma
Statistics from Altmetric.com
Asthma affects up to 30% of the population in westernised societies, with increasing morbidity and costs.1 Rhinoviruses (RVs), the major common cold viruses, are also the most common trigger of asthma exacerbations.2,3 However, the mechanisms by which RVs provoke asthma are not well understood. A number of studies have focused on the interaction of RVs with airway epithelium,4,5 demonstrating the induction of a local inflammatory response. In contrast, information on the immune response to these viruses is limited, though changes in peripheral blood leucocyte counts during and after in vivo RV infection6 suggest that a systemic immune response to RV does develop. The balance between type 1 and type 2 immune responses in CD4 and CD8 T lymphocytes7 and the proposed roles of type 2 responses in the pathogenesis of asthma8 and of type 1 responses in virus infections9 suggest that the nature of immune responses to RV infections in asthmatic and normal subjects may be a subject of major importance in the pathogenesis of virus induced asthma.
Peripheral blood mononuclear cells (PBMC) are activated during RV infection, as indicated by increased production of IL-2 and IFNγ and enhanced NK cytotoxicity after in vitro mitogen stimulation.10 RV16 enters monocytes and airway macrophages in vitro and activates these cells without active replication of the virus,11 inducing non-specific activation of lymphocytes.12 Cytokine responses to RVs in tonsillar cells have been shown to be type 1-like, with large amounts of IL-2 and IFNγ and no IL-4 production.13 These findings are difficult to interpret in the context of allergic asthma, a disease paradigmally connected with type 2 responses. Type 2 responses have been implicated in the pathogenesis of respiratory syncytial virus (RSV) bronchiolitis,14 but responses to RSV have not been examined in asthma.
We have previously reported that RV infections result in bronchial CD4 and CD8 lymphocyte and eosinophil infiltration in both normal and asthmatic subjects.15 Others have also reported eosinophil involvement in rhinovirus infections in atopic/asthmatic subjects.16,17 Our study showed persistent eosinophilia in asthmatic but not normal subjects 6–8 weeks after the RV infection.15 These findings led us to hypothesise that the more severe physiological and inflammatory response observed in asthmatic patients relative to normal subjects may result from differences in lymphocyte function, specifically an imbalance between type 1 and type 2 cytokines.18 The proof of this hypothesis would have major implications in our understanding of the pathogenesis of virus induced asthma. We therefore exposed PBMC from normal and atopic asthmatic subjects to RVs and assessed the secretion patterns of a panel of type 1 and type 2 cytokines. The specificity of these responses was evaluated by inactivating the virus or blocking its cellular receptor.
METHODS
Subjects
Venous blood was drawn from seven subjects with a history of mild to moderate asthma diagnosed by a physician. All were atopic as shown by positive skin prick tests and/or specific IgE to one or more common environmental allergens and a raised total serum IgE (>80 IU/ml). Seven healthy non-atopic subjects were examined in parallel. There were three men and four women in each group and their age distribution was comparable (range 20–57 years). Bronchial provocation with histamine was used to confirm the presence of bronchial hyperresponsiveness in the asthmatic group and the lack of it in the normal subjects. Two asthmatic subjects were receiving low dose inhaled corticosteroids, while the rest of the subjects were not taking any medication at the time of the study. No concomitant diseases were present and no subject had experienced a cold during the previous month. Informed consent was obtained in all cases and the study was approved by the Southampton Hospitals joint ethics subcommittee.
Virus preparations
RV16, a major group subtype,15 was initially obtained from Drs W Busse and E Dick (Madison, WI, USA). RV30, which belongs to the minor RV group, was obtained from the MRC Common Cold Unit (Salisbury, UK). The identity of the viruses was confirmed by neutralisation with specific antisera (ATCC, Rockville, MD, USA). Viruses were propagated in large quantities in Ohio HeLa cells at 33°C in a humidified 5% CO2 incubator and stored at –70°C. For each experiment a new vial was rapidly thawed and used immediately.
RV16 was purified for some experiments to remove soluble factors of HeLa cell origin. An equal volume of saturated ammonium sulphate was added to RV16 suspensions and the virus was precipitated by centrifugation. The precipitant was resuspended in PBS, dialysed overnight at 4°C over PBS using a 50 000 kD MCWO membrane (Sigma Chemical Co, Poole, UK), incubated for 1 hour with 50 μl/ml Staphylococcus protein A-Sepharose Fast Flow (Sigma), clarified, and stored at –70°C.
To study the effect of virus viability and/or receptor binding on the cytokine responses, RV16 was inactivated19 either by (1) exposure to 58°C for 1 hour, (2) exposure to pH 3 for 1 hour at 4°C, or (3) preincubation with 1 mg/ml soluble intercellular adhesion molecule-1 (sICAM-1; kindly donated by P Esmon, Bayer, Berkeley, CA, USA) for 1 hour at room temperature.
Experimental design
PBMC were obtained from whole blood by standard Ficoll centrifugation. They were washed and resuspended in RPMI-1640 medium with Glutamax (Life Technologies, Uxbridge, UK) containing 2.5% FCS and 2.5% human AB serum, without antibiotics, at a final concentration of 2 × 106/ml in 24-well plates. Virus preparations were added at final concentrations ranging from 10 to 0.01 infectious units per cell (multiplicity of infection, MOI). After 1 hour of gentle shaking at room temperature, cultures were placed in a humidified 5% CO2 incubator at 33°C.20 Supernatants were harvested at various time points, clarified by centrifugation, and stored at –70°C until assayed.
Cytokine assays
Levels of IFNγ, IL-12, IL-10, and IL-5 were measured with paired antibodies from R&D Systems (Abingdon, UK) using the manufacturer's recommended concentrations and ELISA protocol. The sensitivity of the assays was 6 pg/ml for IFNγ, 7 pg/ml for IL-12 and IL-10, and 4 pg/ml for IL-5. Commercially available kits were used for the measurement of IL-4 and IL-13 (Biosource, Camarillo, CA, USA). The sensitivities of these assays were <0.27 pg/ml and 12 pg/ml, respectively.
Statistical analysis
Data are expressed as mean (SE). Testing for statistical significance in the time course and dose-response studies was undertaken by analysis of variance (ANOVA). Other comparisons of means were performed using a paired t test.
RESULTS
Cytokine production in PBMC inoculated with RV16
Rhinovirus induction of IFNγ, IL-10 and IL-12 production
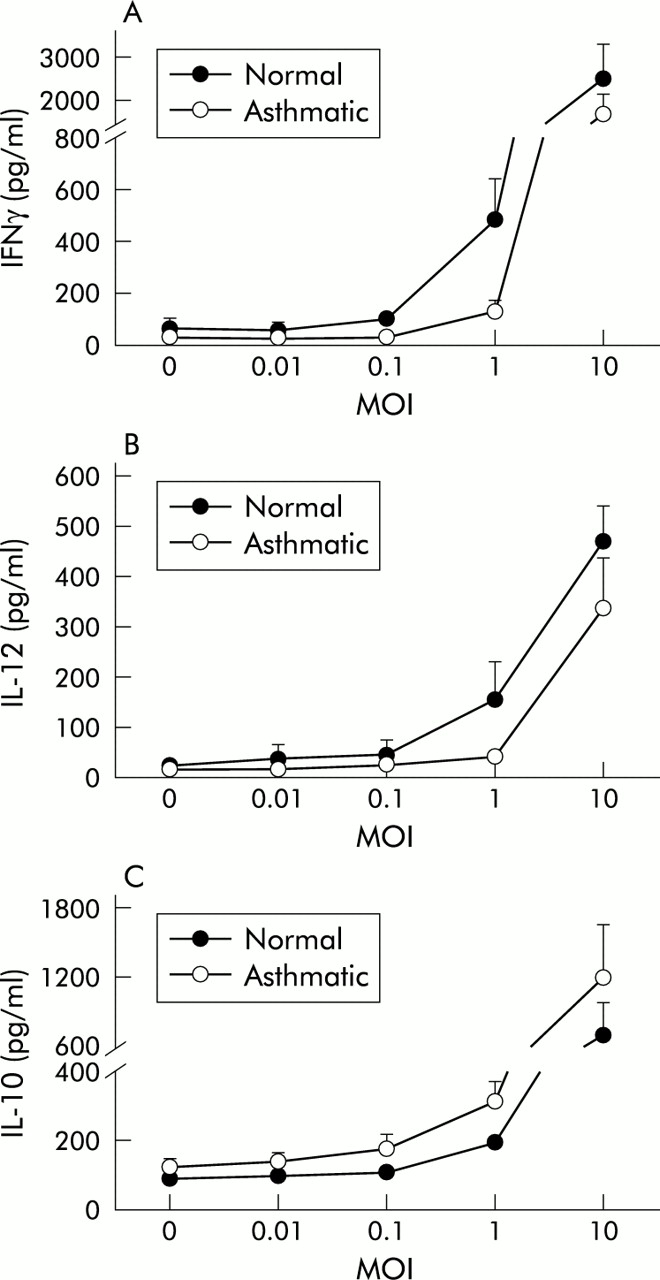
A dose dependent production of IFNγ, IL-10, and IL-12 in response to RV16 inoculation of PBMC was present in both normal and asthmatic subjects with an effective range of 0.1–10 MOI (fig 1). The induction was statistically significant at all concentrations above 0.1 MOI (p<0.05 versus non-infected controls in all cases). Maximal induction occurred with the highest dose studied (10 MOI) (IFNγ: normal 2500 (810) pg/ml, asthmatic 1717 (426) pg/ml; IL-12: normal 469 (72) pg/ml, asthmatic 337 (101) pg/ml; IL-10 normal 695 (277) pg/ml, asthmatic 1199 (442 pg/ml)). A concentration of 1 MOI also produced significant induction and was used in all subsequent experiments.

Dose-response curves of (A) IFNγ, (B) IL-12, and (C) IL-10 production by PBMC in normal (closed symbols) and atopic asthmatic subjects (open symbols) 48 hours after exposure to 0–10 MOI of RV16. Values and error bars are mean (SE). A dose dependent increase in cytokine production was observed for all three cytokines in both groups.
Time course of RV induced IFNγ, IL-10, and IL-12 production
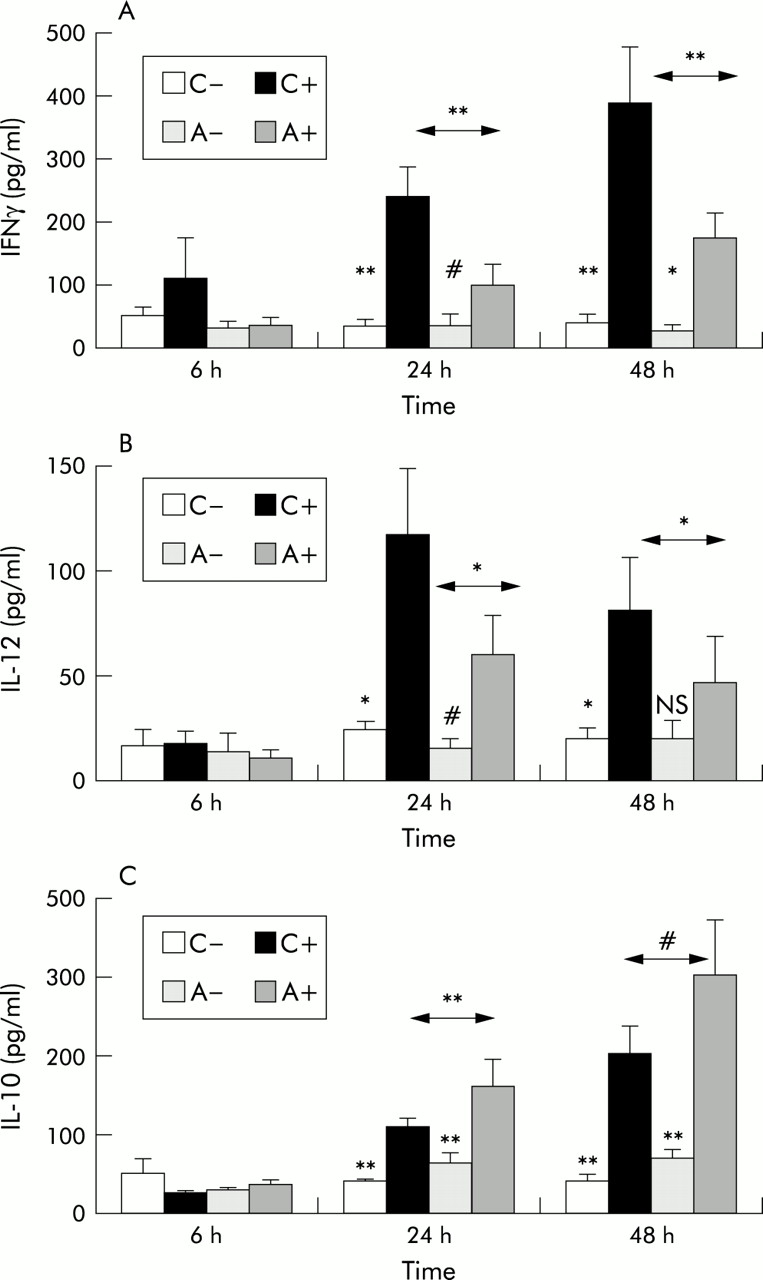
Production of IFNγ, IL-12, and IL-10 in response to RV16 was also time dependent (fig 2). Increasing concentrations of IFNγ and IL-10 were observed at 24 and 48 hours after inoculation, while IL-12 peaked at 24 hours but was still raised at 48 hours. Samples obtained 1 week after inoculation were also available in some experiments. IL-12 was not detectable in those samples, while further increased concentrations of IL-10 were present in the RV infected samples and significant amounts of IFNγ still remained (not shown), suggesting that RV mediated activation persisted in these cultures.

Time course of (A) IFNγ, (B) IL-12, and (C) IL-10 production by PBMC of normal and atopic asthmatic subjects 6–48 hours after exposure to 1 MOI of RV16 or medium alone. C– = non-infected cultures from normal subjects, C+ = RV16 infected cultures from normal subjects, A– = non-infected cultures from subjects with atopic asthma, A+ = infected cultures from subjects with atopic asthma. Values are mean (SE). Analysis of variance (ANOVA) was used to compare within and between group values at each time point. Exposure to RV16 resulted in a significant induction of IFNγ, IL-12, and IL-10 production 24 hours after inoculation, increasing further at 48 hours for IFNγ and IL-10. Although this effect was observed in both normal subjects and those with atopic asthma, significant differences were observed between the groups with lower levels of IFNγ and IL-12 and higher levels of IL-10 in asthmatic patients than in normal subjects. **p<0.01, *p<0.05, #0.05<p<0.1, NS = non-significant.
Differences in cytokine induction by RV between normal and atopic asthmatic subjects
Production of IFNγ from PBMC of normal subjects after exposure to RV16 was significantly higher than in asthmatic subjects at both 24 hours (normal subjects 239 (46) pg/ml, asthmatic subjects 99 (31) pg/ml, p<0.01) and 48 hours (normal subjects 386 (91) pg/ml, asthmatic subjects 175 (39) pg/ml, p<0.01; fig 2A). IL-12 was also significantly higher in normal subjects than in those with atopic asthma 24 hours (normal subjects 115 (31) pg/ml, asthmatic subjects 58 (19) pg/ml, p<0.05) and 48 hours after inoculation (normal subjects 80 (25) pg/ml, asthmatic subjects 46 (21) pg/ml, p<0.05; fig 2B). In contrast, levels of IL-10 (fig 2C) were significantly higher in cultures from asthmatic subjects than in normal subjects at 24 hours (normal subjects 108 (11) pg/ml, asthmatic subjects 159 (35) pg/ml, p<0.05) and marginally higher at 48 hours (normal subjects 200 (25) pg/ml, asthmatic subjects 298 (69) pg/ml, 0.05<p<0.1). All these differences were also consistently present in the dose-response experiments (fig 1).
RV induced production of type 2 cytokines from PBMC
Very low levels of IL-4 were detectable in the cultures (fig 3A). No RV mediated IL-4 upregulation was observed in supernatants of PBMC from normal subjects. In contrast, IL-4 levels in supernatants from RV stimulated cells from asthmatic subjects 24 hours after inoculation were higher than in those from non-stimulated cultures (820 (257) fg/ml v 268 (64) fg/ml; 0.05<p<0.1). Furthermore, to investigate the balance in type 1 and type 2 cytokine production, IFNγ and IL-4 levels were expressed as ratios and compared. The ratio of IFNγ to IL-4 production in response to RV at 24 hours was more than three times higher in normal subjects (938 (246)) than in those with asthma (267 (93), p=0.028, fig 3B). RV16 inoculation also induced a moderate upregulation of IL-13 (control 143.6 (82.5) pg/ml, RV16 360.2 (129.2) pg/ml, p<0.05), with no differences between the normal and asthmatic subjects. No virus mediated induction of IL-5 or any differences between the groups were observed (not shown).

Rhinovirus mediated effects on type 2 cytokine production by PBMC from normal and atopic asthmatic subjects. (A) IL-4 production in normal subjects did not differ between non-infected (C–) and RV16 infected (C+) cultures. A marginal induction of IL-4 was observed 24 hours and 48 hours after exposure of PBMC from asthmatic subjects to 1 MOI of RV16 (A+) in comparison with the control cultures (A–; 0.05<p<0.1, ANOVA). (B) Ratio of IFNγ to IL-4 production 24 hours after exposure to 1 MOI of RV16 was more than threefold higher in cultures from normal subjects than in those with atopic asthma (p=0.028, t test). *p<0.05, #0.05<p<0.1.
Virus specificity and serotype and receptor specificities of cytokine induction
To determine whether the responses observed were specific to RV rather than a response to soluble factors in the virus inoculum, we examined the responses to purified virus. When purified RV16 was compared with non-purified virus preparations no differences were observed in the production of IL-10 (182 (24) pg/ml v 168 (42) pg/ml), IL-12 (110 (32) pg/ml v 84 (20) pg/ml), or IFNγ (301 (52) pg/ml v 361 (77) pg/ml, n=5; p>0.1 in all cases).
Inoculations with RV30, a minor group serotype,21 at 1 MOI in parallel cultures resulted in IFNγ production at the same level as RV16 inoculation (372 (81) pg/ml, n=3, p>0.1), indicating that cytokine induction by PBMC in response to RV is not confined to a specific RV serotype. These results also exclude the possibility that the observed induction of IFNγ in response to RV16 was restricted through signalling via ICAM-1, the receptor of RV16, but not of RV30, as similar levels of induction were observed with both serotypes.
Effect of virus inactivation and receptor blockade
Exposure to high temperature or acid pH completely inactivated RV16, as assessed by titration assays on Ohio-HeLa cells (not shown). These pretreatments were also able to markedly inhibit RV16 mediated induction of IFNγ in PBMC (fig 4A), indicating that live virus was necessary to induce the major part of IFNγ production. In contrast, the same pretreatments resulted in only partial reduction of RV mediated IL-10 production, the reductions being non-significant for acid treatment and statistically marginal for heat inactivation (p=0.088, fig 4B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of virus inactivation on production of (A) IFNγ and (B) IL-10 by PBMC exposed to RV16. To inactivate the virus RV16 was exposed to temperatures of 58°C, pH 3, or sICAM-1. IFNγ production was completely abolished by all inactivating treatments (p<0.05 v infectious virus preparation in all cases, ANOVA). Only small decreases in RV16 mediated IL-10 production were observed in the same cultures (temperature: 0.05<p<0.1; pH: p=NS; sICAM: p<0.05 v infectious virus preparation).
Similar to the above experiments, blocking RV16 receptor binding sites by precoating the virus with sICAM-1 resulted in marked inhibition of RV mediated IFNγ upregulation (p<0.05 v non-inactivated positive control, fig 4A). Again, RV induced production of IL-10 was much less affected by receptor blockade, although a statistically significant reduction was observed (p<0.05, fig 4B). No differences between normal and asthmatic subjects were detected in any of the inactivation experiments.
These data indicate that the major part of IFNγ production by PBMC in response to RV requires both virus receptor binding and virus capable of active replication. In contrast, IL-10 production by PBMC in response to RV is to some extent independent of both virus receptor binding and the capacity of the virus to replicate.
DISCUSSION
Asthma is increasingly common and RV infections are the commonest trigger of asthma exacerbations.22 Despite this, very little is known about the host response to this virus and its potential implications in the mechanisms inducing asthma. Several novel findings in this respect are reported in this study. Firstly, exposure of PBMC to RVs resulted in a time and dose dependent upregulation of IFNγ, IL-12, and IL-10 production in both normal and atopic asthmatic subjects. IL-13 was moderately induced in both groups, while IL-4 was induced only in the atopic asthmatic group. A finding of major importance was the deficient production of the type 1 cytokines IFNγ and IL-12 in the asthmatic group compared with the normal subjects.
Of these mediators, IFNγ has previously been studied in the context of RV infections12,13 where it was found that its induction did not require cell contact but was mediated by monocyte derived soluble factor(s).12 Among cytokines able to mediate such an effect, IFNα is highly inducible after RV infection23; however, no information was previously available with regard to IL-12. While IL-12 is considered a pivotal cytokine for promoting NK and Th1 responses after bacterial or parasite infections,24 its role in antiviral immunity is less clear; there is evidence that in some viral infections IFNγ responses may be IL-12 independent.25 It has recently been proposed that IFNα and IL-12 can be induced simultaneously; either will lead to IFNγ production but conditions of high IFNα expression would inhibit the IL-12 response to a different extent, depending on the specific viral pathogen.26 Our data are compatible with this hypothesis: IL-12 production peaked 24 hours after RV inoculation and declined thereafter, while IFNα is known to peak 48–72 hours after RV infection.23 The presence of high concentrations of IFNα 48 hours after infection was also confirmed in our samples (data not shown).
IL-12 and IFNγ are generally believed to control rather than induce asthma related pathophysiology, even though the ability of IFNγ to prolong eosinophil survival and stimulate pro-inflammatory activities are potential pathways through which this cytokine may contribute to allergic inflammation.27 Nevertheless, our findings show for the first time that the normal type 1-like antiviral response is defective in subjects with atopic asthma, both at the inducing (IL-12) and effector (IFNγ) levels.
Furthermore, in contrast to previous observations,13 there is an induction of type 2 cytokines after exposure to RVs. This effect is less prominent than the dominant type 1 response, but it could be important in the context of asthma exacerbations, especially as it complements recent evidence suggesting type 2 cytokine involvement in animal models of RSV and other virus infections.28,29 Levels of IL-4 were very low, which is probably the reason why previous attempts to detect it were negative.13 Nevertheless, RV mediated IL-4 upregulation was observed in the atopic asthmatic group. This is consistent with our understanding of the reciprocal regulation of type 1 and type 2 responses 22: IFNγ concentrations almost three times higher than IL-4 in normal subjects may have completely suppressed IL-4 induction, while this ratio was more than three times lower in the atopic asthmatic group (fig 3B). IL-13, a cytokine critical to allergen induced asthma,30 was upregulated by RVs in both normal and asthmatic subjects. IL-13 can induce IgE production independently from IL-4, but it can also induce asthma through non-IgE dependent mechanisms.31 Finally, IL-10 was significantly induced in both groups, predominating in asthmatic subjects. The induction of IL-10 in macrophages32 or PBMC33 has also been reported after respiratory syncytial virus infection. It has been suggested32 that the inhibitory activities of IL-10 could lead to an ineffective immune response against that virus; however, the role of this cytokine in asthma is debatable as both increased34 and decreased35 levels have been reported in different settings.
Further interesting observations were obtained from the inactivation experiments. RV mediated IFNγ upregulation was almost completely blocked by all the different approaches that prohibit virus receptor binding and/or viral replication in cells (temperature, acid inactivation of the virus, and receptor blockade by sICAM-1). This is compatible with a previous report by Gern et al showing complete inhibition of RV mediated CD69 induction on T cells by ICAM-1 blockade.12 Moreover, these data and the identical IFNγ responses obtained with purified and non-purified virus preparations prove that the response is RV specific and is not contributed to by HeLa cell derived factors. In contrast, IL-10 production was only partly affected by the inactivation treatments. However, IL-10 production did not differ between purified and conventional RV preparations, suggesting that there were no other IL-10 inducing factors in the latter. Interestingly, these results are in agreement with another study11 in which RV mediated TNFα upregulation could not be blocked with ultraviolet inactivation of the virus. Both IL-10 and TNFα are products of monocytes/macrophages; the lack of inhibition of RV binding to macrophages by ICAM-1 blockade observed in that study11 led the authors to attribute these findings to either an additional RV receptor on some cells or a non-specific entry of RV into monocytes (for example, by phagocytosis). Additional evidence supporting such a possibility can be found in reports of RV strains resistant to ICAM-1 inactivation in epithelial cells.36 Although we find these suggestions intriguing, further detailed studies are needed to understand fully the mechanisms of IL-10 induction.
Overall, the above data suggest that the host response to RVs is more complicated than previously reported, involving both type 1 and type 2 elements. Furthermore, this response is aberrant in atopic asthmatic subjects in a manner similar, but not identical, to their response to allergens. This is the first time that such impairment has been shown for RV infections or any other respiratory pathogen in a human model. As it is well established that type 1 cytokines are necessary for the clearance of the virus9—in contrast to type 2 cytokines which delay virus clearance37—persistence of the viral stimulus could lead to augmentation of the inflammation which characterises asthma. Replication of RV in PBMC is either absent or of a very low grade, but the virus might persist in cells for a prolonged period.38
We have previously demonstrated a significant influx of CD4 and CD8 lymphocytes into the bronchial mucosa after RV infection.15 In a milieu with increased type 2 cytokines, as in the asthmatic lung, a further shift towards type 2 responses may occur.29 It is therefore plausible that a defective type 1 immune response to RV in predisposed atopic asthmatic individuals may significantly contribute to the development of acute exacerbations of asthma by promoting type 2 inflammation associated with allergic/asthmatic disease and diminishing viral clearance through deficient type 1 antiviral immune responses. This hypothesis is supported by recent data from a study of patients with allergic rhinitis or asthma experimentally infected with RV16 in whom virus induced changes in the sputum type 1/type 2 cytokine ratio were inversely related to both peak cold symptoms and time to viral clearance.39 Further studies are now required to investigate this hypothesis in asthmatic and normal subjects.