Article Text

Statistics from Altmetric.com
Amyloidosis is a generic term for a heterogeneous group of disorders associated with deposition of protein in an abnormal fibrillar form.1 The diverse spectrum of amyloid related diseases is now recognised to include Alzheimer’s disease, type II diabetes, and the transmissible spongiform encephalopathies. Amyloidosis can be hereditary or acquired, localised or systemic, and potentially lethal or merely an incidental finding.
Amyloid deposits consist mainly of protein fibrils, the varying peptide subunits of which constitute the basis for its classification (table1).2 Despite much heterogeneity among their respective precursor proteins, all amyloid fibrils have a remarkably similar adopted ultrastructure and share many physicochemical properties attributable to their acquired rich β-sheet content.3Certain glycosaminoglycans (GAGs) are invariably associated with the fibrils and, in addition, all amyloid deposits contain the normal plasma protein serum amyloid P component (SAP). The specific binding interaction between SAP and all amyloid fibrils is the basis for our development of radiolabelled SAP as a diagnostic nuclear medicine tracer.4 ,5
Classification of the more common types of amyloid and amyloidosis
There have been many recent advances in amyloidosis including elucidation of the structure and properties of amyloid fibrils, the role of GAGs and SAP, and substantial improvements in clinical diagnosis and management. Although a specific generic treatment for the disease is not yet available, compelling evidence that amyloid deposits frequently regress when the supply of the respective fibril precursor protein is reduced has encouraged a much more positive approach to patient care.5
Pathogenesis of amyloidosis
Amyloidosis is a disorder of protein folding which breaks the traditional dogma that amino acid sequence is the sole determinant of a protein’s tertiary form. Amyloid fibril proteins can evidently exist as two radically different stable structures—a normal soluble form and the highly abnormal fibril conformation.
The pathogenesis of amyloidosis thus centres around “off pathway” folding of the various fibril precursor proteins into an alternative conformation rich in β-sheet structure which can form the characteristic fibrils by auto-aggregating in a highly ordered fashion.6 Fibril diffraction studies have confirmed that the β-strands within the β-sheets are arranged specifically, and that all amyloid fibrils share an essentially similar core structure.7 ,8 This underlies their distinctive physicochemical properties including their ability to bind Congo red in a spatially organised manner,9 relative resistance to proteolysis, and capacity to bind SAP.1 Most fibril precursor proteins will form amyloid fibrils in vitro and in some instances the fibrils in vivo are composed of intact whole precursor molecules—for example, variants of transthyretin (TTR) and lysozyme in hereditary amyloidosis and β2-microglobulin in dialysis related amyloidosis (DRA).10-12 More often the precursor proteins undergo partial cleavage, although it is not known exactly when this occurs.13 ,14
Although the amino acid sequence underlies the potential for a protein to form amyloid, and an essential prerequisite for its development is a sustained supply of the respective fibril precursor, little is known about the genetic or environmental factors that determine individual susceptibility to amyloid, or those which govern its anatomical distribution and clinical effects. For example, only a small proportion of patients with chronic inflammatory disease develop AA amyloidosis, and can do so at any time from one year to many decades after acquiring the underlying disorder.15 However, parenteral injection of a minute extract of amyloidotic tissue primes mice for explosively rapid AA amyloid deposition within days of a single brief acute phase stimulus, and although the significance of this so called “amyloid enhancing factor” (AEF)16 has long been appreciated, it remains surprisingly poorly characterised. AEF evidently promotes amyloidogenic off pathway folding of susceptible proteins, possibly by capturing them on to an “amyloid template” when they transiently populate unstable partly unfolded forms. Since amyloid is massively rich in AEF, the conditions necessary for amyloid formation in vivo are probably self-perpetuating once an initial nucleus of amyloid material has been laid down, depending only on continued supply of the fibril precursor protein. It is of interest that some normal tissues contain low levels of AEF,17raising the possibility that proteins may quite commonly be processed transiently in an amyloid-like manner.
The universal presence in amyloid of certain GAGs, notably heparan and dermatan sulphate, and SAP has long suggested that these moieties might contribute to its pathogenesis. The role of SAP has lately been confirmed in SAP “knock-out” mice in which experimentally induced AA amyloidosis was substantially reduced.18 Similarly, there is indirect evidence that the fibril associated GAGs enhance amyloidogenesis.19 Although neither SAP nor GAGs are required for amyloid fibrillogenesis per se, they both remain valid therapeutic targets.20
Many of the pathological effects of amyloid can be attributed to its physical presence. The deposits accumulate in the extracellular space, progressively disrupting tissue architecture, and can both impair organ function and produce space occupying effects. Amyloid fibrils may also be cytotoxic, possibly by promoting apoptosis,21 which could account for their damaging consequences in Alzheimer’s disease in which the deposits are scanty.
The typically progressive nature of amyloidosis has given rise to the notion that amyloid is inert and irreversible, although in reality this more closely reflects the unremitting character of the various conditions which underlie it. Regression of amyloid has been described in numerous case reports, and systematic radiolabelled SAP scans in over 1000 patients (see below) have lately confirmed that the deposits can regress quite rapidly when the supply of fibril precursors is reduced. Although amyloid deposits are unusually stable, they do turn over continuously in vivo, albeit at a low and variable rate which is readily exceeded by the rate of deposition.
HEREDITARY AMYLOIDOSIS AS A MODEL OF PATHOGENESIS
Genetic variants of certain proteins such as TTR and lysozyme, form amyloid fibrils very readily and cause distinct autosomal dominant hereditary amyloidosis syndromes. Although hereditary amyloidosis is extraordinarily rare, it represents an invaluable model for studying the pathogenesis of amyloidosis generally. At the molecular level, genetically variant proteins with a strong propensity to form amyloid can be compared with their wild-type counterparts. Variants of TTR have been studied most widely, but comparison with the wild-type protein, which forms fibrils in senile systemic amyloidosis,22 is not ideal. The recent discovery of hereditary systemic amyloidosis caused by human lysozyme mutations12 promised much since, like TTR, its complete three dimensional structure was known, but wild-type lysozyme is not associated with amyloidosis.
The amyloidogenic lysozyme variants Thr56 and His67 proved to be less stable than wild-type and can form amyloid fibrils in vitro either following heating or prolonged standing at 4°C.12 These variants can unfold partially in vitro whilst retaining secondary structure, suggesting they are able to adopt a molten globule intermediate form (fig 1). It is thought that both mutations destabilise lysozyme sufficiently for this to happen transiently in vivo enabling intermediates, which retain major elements of the β-structure of the native fold, to restabilise themselves by intermolecular aggregation and thereby producing the cross-β fold typical of amyloid fibrils. Remarkably, this process is completely reversible since fully functional soluble lysozyme His67 can be recovered in vitro from isolated ex vivo amyloid fibrils under specific conditions.3 Elucidation of the intermolecular β-sheet association that occurs between pre-fibril intermediates may assist the development of generic fibrillogenesis inhibitors.
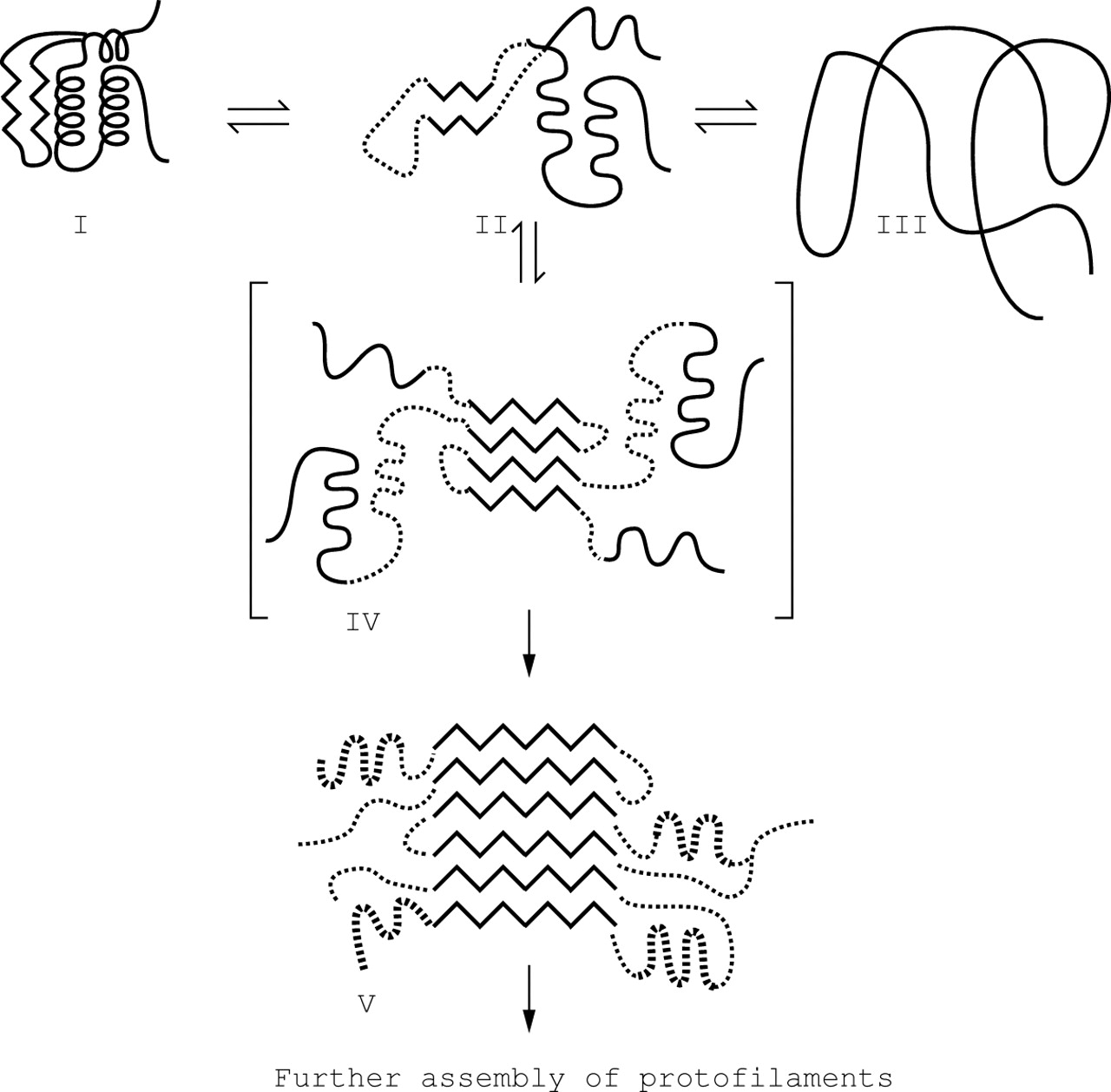
Proposed mechanism for lysozyme amyloid fibril formation. A partly folded, molten globule-like form of the protein (II), distinct from the native (I) and denatured (III) states, self-associates through the β-domain (IV) to initiate fibril formation. This provides the template for further deposition of protein and for the development of the stable, mainly β-sheet, core structure of the fibril (V). Reprinted with permission from Booth et al, Nature 1997;385:787–793.Copyright (1997) Macmillan Magazines Ltd.
SPECTRUM OF AL AMYLOIDOSIS
AL amyloidosis is the most heterogeneous form of the disease, consistent with the fibril protein being unique in each patient. Virtually any organ system excluding the brain can be involved, and in almost any combination. The clonal B cell diseases that underlie AL amyloid also vary enormously in that they can be benign or malignant, localised or generalised, and relatively chemosensitive or refractory to treatment.
Localised AL amyloidosis is most often identified in the upper respiratory, urogenital and gastrointestinal tracts, the skin, and orbit.1 In such circumstances the amyloidogenic light chains are produced by a subtle clone of lymphoplasmacytes that is localised in proximity to the amyloid deposits.8 ,23 ,24This type of amyloid is frequently nodular in character, but can occur quite diffusely throughout a particular tissue when it is associated with a more contiguous infiltrate of clonal cells.25 ,26The AL nature of localised amyloid can often be confirmed immunohistochemically or by sequencing the fibril protein, but it may not be possible to characterise the associated clonal cells due to their scanty nature.
In contrast, the fibrils in systemic AL amyloidosis are derived from circulating monoclonal light chains that are usually produced by a low grade intramedullary clonal plasma cell dyscrasia. Although organ involvement in systemic AL amyloidosis is extremely heterogeneous, the deposits are widespread in every case. Even when the function of only a single organ is demonstrably impaired, there is always diffuse involvement of blood vessels and occult deposits in other organs frequently become significant under the stress of surgery, infections, or failure of another system.
Respiratory tract amyloidosis
CLASSIFICATION
Amyloid localised to the respiratory tract was recognised by Lesser in 1877, since when various classifications have been proposed based upon radiographic or bronchoscopic findings.27 ,28Although amyloidosis is now formally classified by fibril protein (table 1),2 there are some characteristic clinical syndromes in respiratory amyloidosis that point strongly to their molecular basis (table 2). Inclusion of pulmonary vascular amyloidosis as a clinical syndrome is confusing since this is a histological finding that occurs to some extent in all its systemic forms.
Respiratory tract amyloidosis syndromes
There have been a few reports of systemic AA amyloidosis affecting the lungs but fibril typing has generally been imperfect29-31and all studies in which the fibril protein has been sequenced identified AL type.32-34 Respiratory tract involvement has not been a clinical feature in over 200 patients with systemic AA amyloidosis evaluated in our unit and, likewise, pulmonary TTR amyloid occurs histologically in senile systemic amyloidosis but its clinical significance is doubtful.29 ,35 Prominent lung disease is not a recognised feature of hereditary amyloidosis. In most situations, therefore, respiratory amyloidosis will be of AL type although the presence of chronic inflammatory disease or a family history or extreme old age should signal other possibilities.
LARYNGEAL AMYLOIDOSIS
Amyloid represents 0.5–1% of benign laryngeal disease, increasing with age but occasionally affecting young adults.36 Symptomatic laryngeal amyloidosis is usually localised but rarely can be a manifestation of systemic AL type.37 ,38 Discrete nodular and diffuse infiltrative types of laryngeal amyloid were described in 1919,39 the diffuse pattern with an intact mucosa being more common, sometimes with tracheobronchial extension.37 Symptomatic laryngeal amyloidosis is usually localised but can rarely be a manifestation of systemic AL type.37 ,38 The amyloid deposits occur most commonly in the supraglottic larynx and usually present with hoarseness or stridor, but can cause a sensation of “fullness” in the throat, choking, and dyspnoea on exertion.37 The disorder is usually relatively benign but can be progressive or recur after treatment. Fatal haemorrhage has been reported.40
TRACHEOBRONCHIAL AMYLOIDOSIS
Tracheobronchial amyloidosis is an uncommon diagnosis.27 ,41 A literature review in 1983 identified only 67 cases worldwide, of which 57 were diffusely infiltrative (multifocal submucosal plaques) and the remainder were nodular or “tumour-like”.28 The disorder has been associated with tracheobronchopathia osteoplastica, which is characterised by calcified or cartilaginous submucosal nodules within the airways.42-44
Tracheobronchial amyloidosis typically presents after the fifth decade with dyspnoea, cough and, occasionally, haemoptysis. Narrowing of the airways can cause distal atelectasis or recurrent pneumonia, and solitary nodules may be mistaken for endobronchial neoplasia.45 Although histological involvement of airways is probably frequent in systemic AL amyloidosis,46symptomatic tracheobronchial amyloidosis is usually localised. However, its course is not necessarily benign: three of seven cases followed up by Hui died of respiratory failure or secondary pneumonia,30 and three of four patients at the Mayo Clinic died within 79 months of diagnosis.27 In contrast, three of four patients followed up by Cordier for a median of eight years remained well.41
PARENCHYMAL AMYLOIDOSIS
Amyloid involving the lung parenchyma is the most frequently diagnosed respiratory amyloidosis syndrome. It remains useful to distinguish it radiographically into solitary/multiple nodules or a diffuse alveolar septal pattern since the former is usually localised AL type and the latter usually a manifestation of systemic amyloidosis. In one series of 48 patients with localised respiratory tract amyloidosis, 34 had parenchymal deposits of which 28 cases were nodular and six were diffuse alveolar septal.30 None of the latter were reported in a similar series of 17 patients from the Mayo Clinic27 and Cordier found a systemic (AL) aetiology in the majority of 15 patients with diffuse alveolar septal amyloidosis.41
Amyloid nodules in the lung parenchyma are usually an incidental finding that need to be distinguished from neoplasia. They are usually peripheral and subpleural, occur more frequently in the lower lobes, may be bilateral, and range in diameter from 0.4 cm to 15 cm (fig 2). They grow slowly and frequently cavitate or calcify.47-49Larger nodules can occasionally produce space occupying effects.
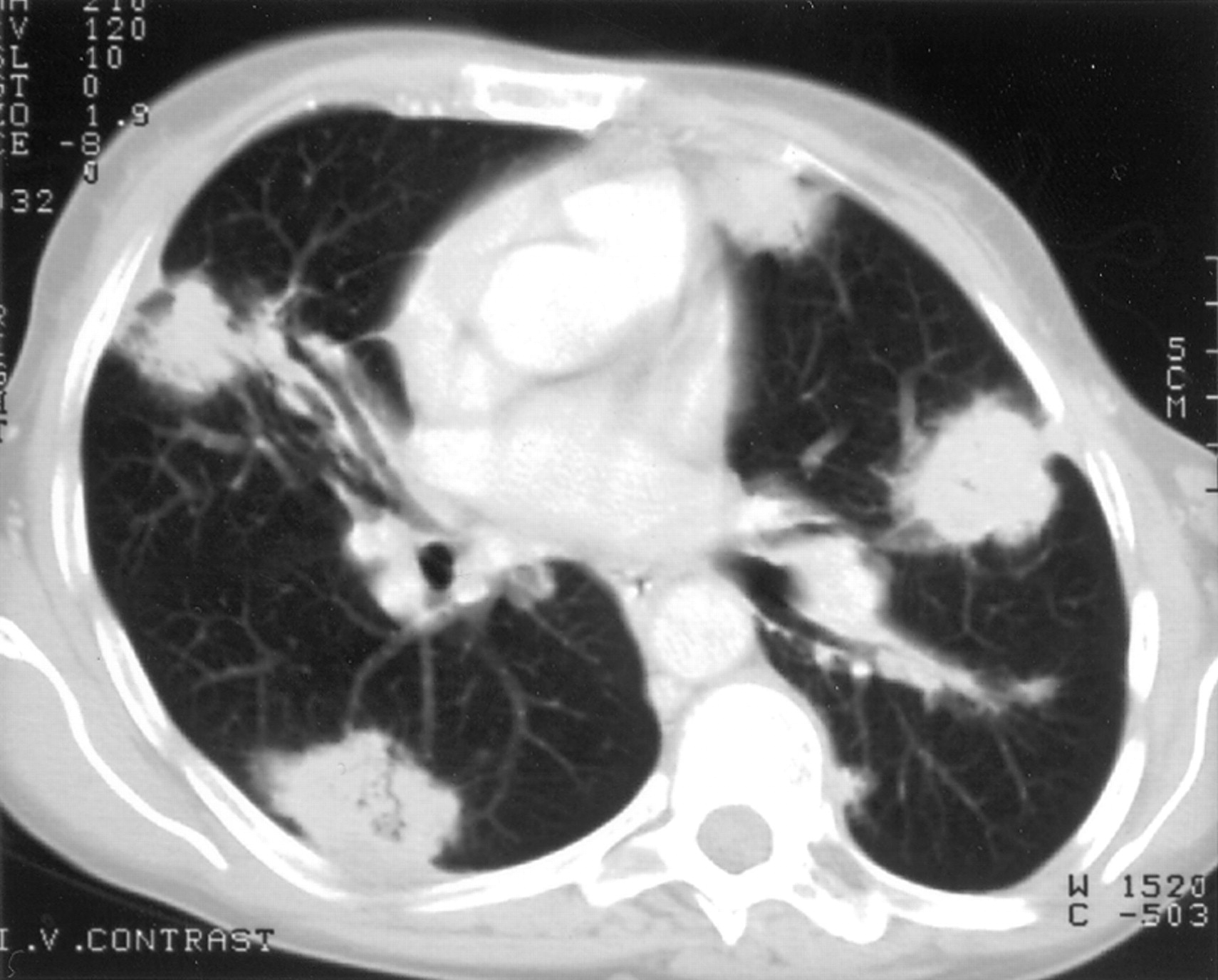
Nodular parenchymal amyloidosis. CT scan of a patient showing multiple, bilateral, subpleural soft tissue densities, histological analysis of which confirmed the presence of amyloid.
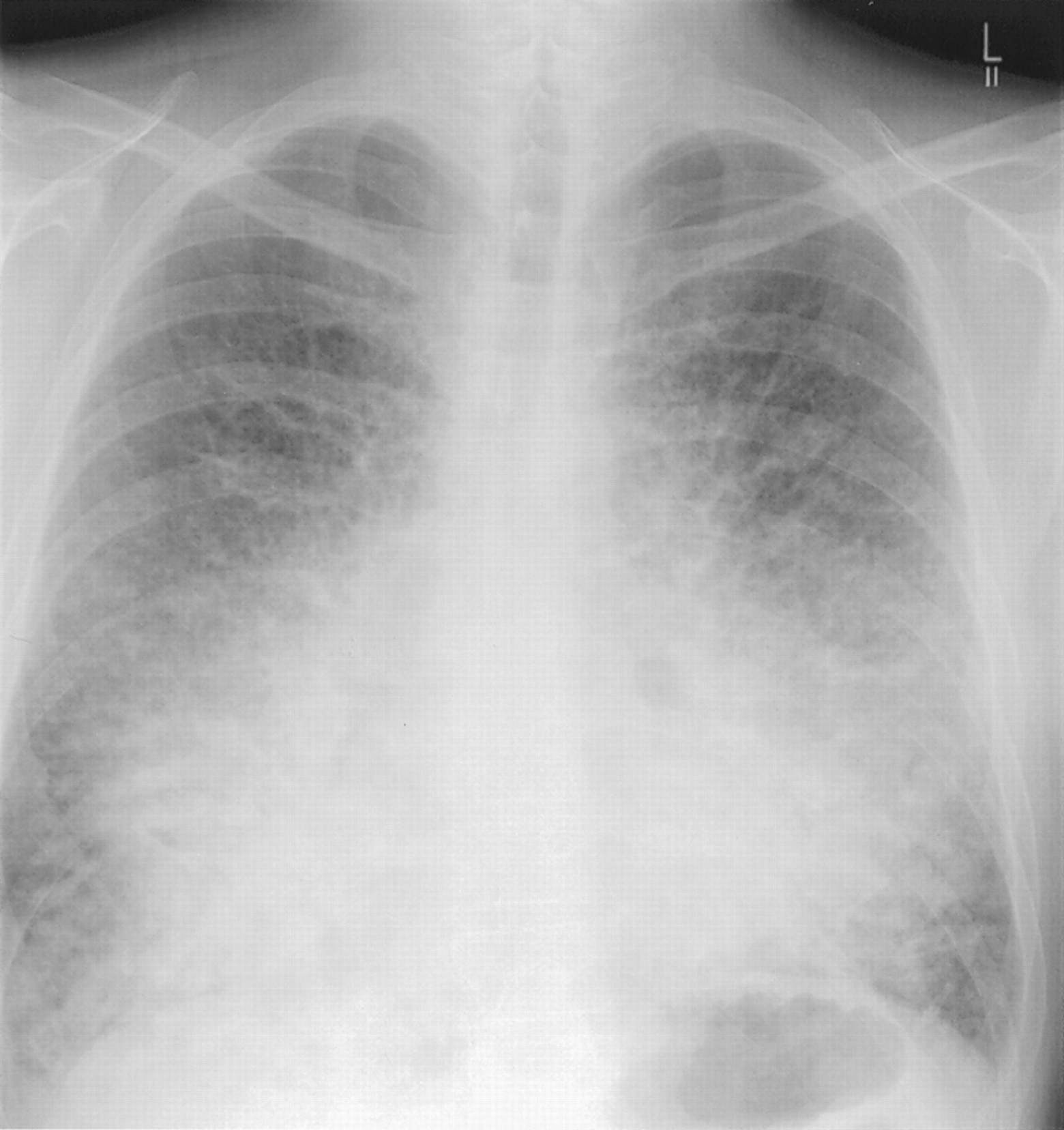
Autopsy series have confirmed that diffuse parenchymal amyloid is common histologically in systemic AL amyloidosis.29 ,46 Clinical involvement is rare but can be confused with pulmonary oedema and/or fibrosis (fig 3). Signs are often scanty. Respiratory function tests may, but not always, reveal a restrictive defect with impaired gas exchange,41but it can be difficult to determine the relative contribution to symptoms of pulmonary versus cardiac amyloid which frequently co-exist.29 Pulmonary involvement is not a major contributor to death in AL amyloidosis41 ,46 and the median survival of patients with clinically overt lung deposition is about 16 months, similar to that for systemic AL amyloidosis generally.27

Systemic AL amyloidosis with diffuse alveolar septal parenchymal lung involvement. Digital chest radiograph of a patient who presented with dyspnoea and cough showing diffuse interstitial shadowing causing loss of definition of the cardiac borders. A bronchoscopic biopsy specimen revealed amyloid and bone marrow examination revealed a monoclonal lymphoplasmacytoid population.
MEDIASTINAL AND HILAR AMYLOIDOSIS
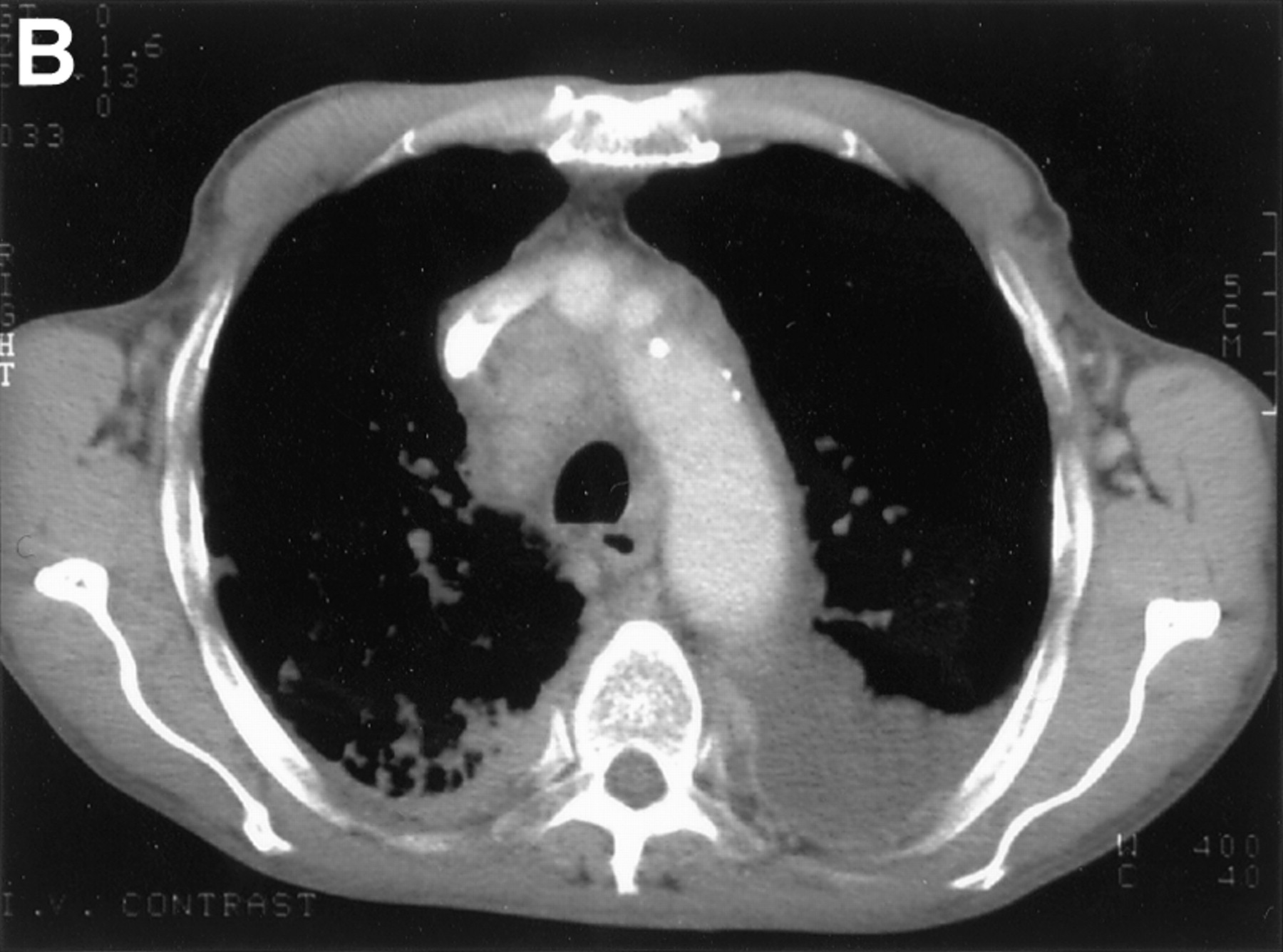
The lymphatic system is frequently affected in systemic AL amyloidosis although predominant lymph node deposition is unusual (fig4).50 ,51 Hilar or mediastinal adenopathy is rarely associated with localised pulmonary amyloidosis28 ,52 and its discovery should prompt a search for a systemic aetiology. Amyloid lymphadenopathy can also represent localised AL deposition in association with solitary or multifocal B cell lymphomas.

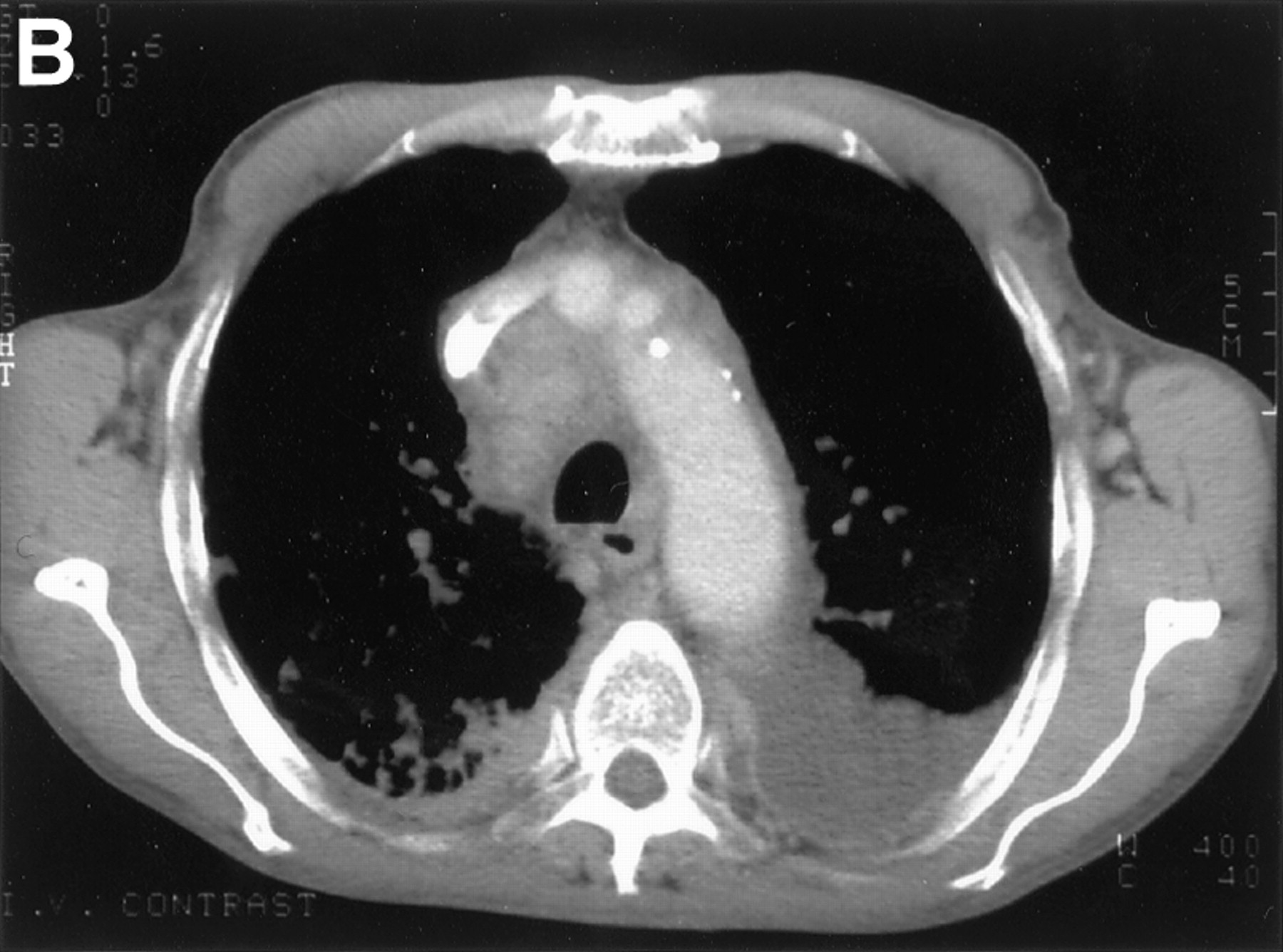
(A) Plain radiograph and (B) thoracic CT scan of a patient who presented with generalised lymphadenopathy and progressive dyspnoea revealing paratracheal and hilar lymphadenopathy associated with diffuse lung infiltration and pleural effusions. Histological examination of the lymph nodes confirmed AL amyloidosis.
Hilar adenopathy can be unilateral or bilateral and may calcify.53 ,54 Although usually asymptomatic, there have been occasional reports of tracheal compression and superior vena caval obstruction.
Diagnosis
The diagnosis of amyloidosis usually requires histological confirmation, and Congo red staining that produces green birefringence under crossed polarised light remains the gold standard.55Most tissue specimens, ranging from needle biopsies to open surgical resections,27 can be studied for amyloid although small biopsy specimens are open to significant sampling error. Furthermore, false positive results do occur in routine practice, usually due to poor Congo red methodology. Amyloidotic tissues can bleed following biopsy sampling and, although this is unlikely after fine needle aspiration,56-58 the possibility should be considered when endobronchial and transbronchial biopsy specimens are taken.27
Positive histology results for amyloid must be followed up by immunohistochemical analysis to determine the fibril type. Suitable antibodies are widely available but unfortunately AL deposits often do not stain, presumably because these fibrils are derived from variable domain monoclonal light chain fragments that are unique in each case.59 However, immunohistochemical exclusion of AA and ATTR makes a diagnosis of AL type overwhelmingly likely.
Anatomical and functional evaluation of the tissues/organs affected by amyloid need to be tailored to each case and, in amyloidosis of the respiratory tract, are likely to include plain radiography, CT scanning, endoscopy, and comprehensive respiratory function tests. Evidence of systemic disease should be sought clinically, including urinalysis, and by performing haematological and biochemical profiles. The intramedullary clone of plasma cells that underlies systemic AL amyloidosis is typically very subtle and should be sought by bone marrow examination and immunofixation of serum and urine for a monoclonal protein. Immunoglobulin gene rearrangement studies may identify subtle clones in either the bone marrow or, in the case of localised AL, within the amyloidotic (respiratory tract) tissue itself.60
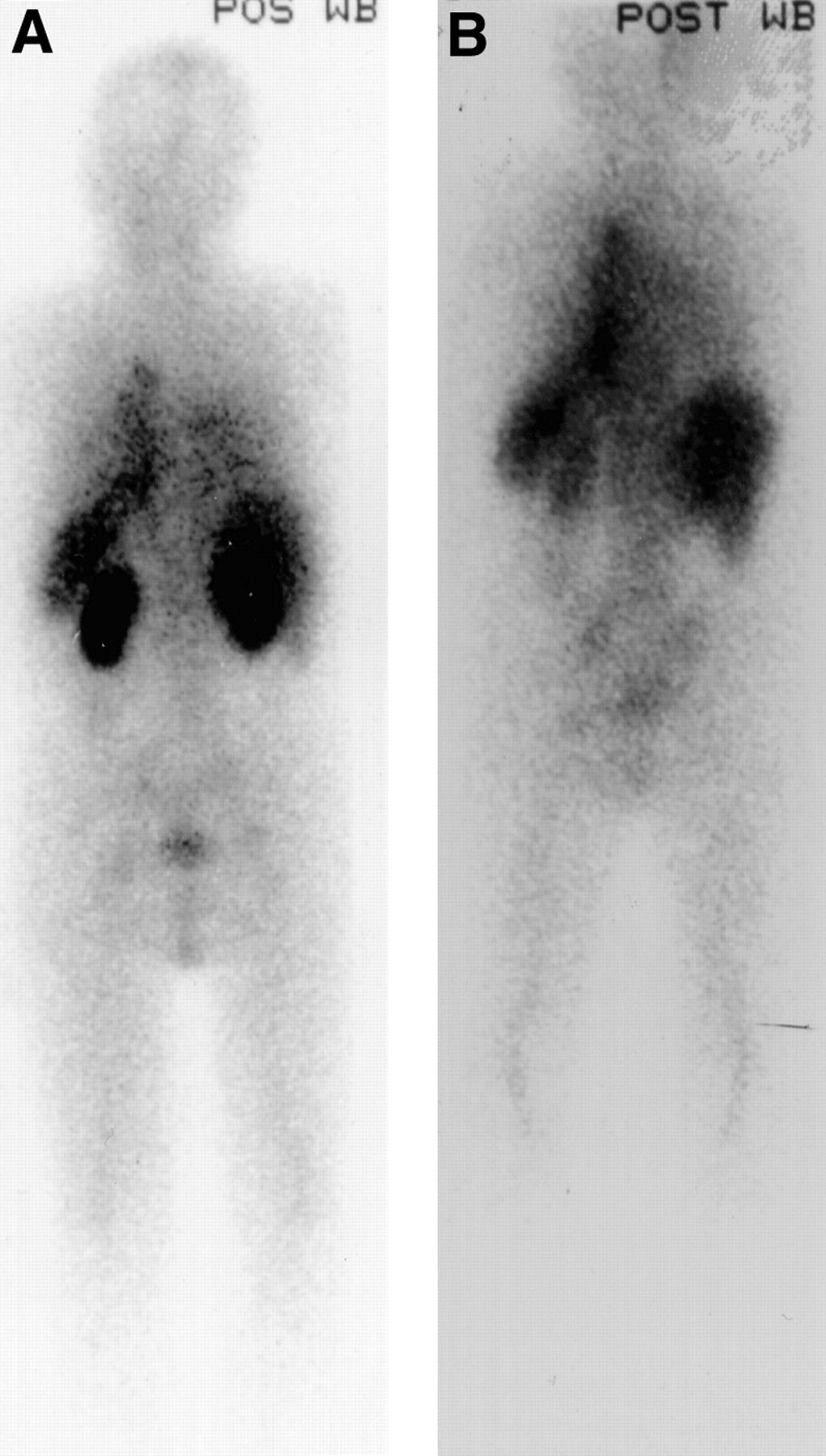
Radiolabelled SAP localises specifically to amyloid deposits in vivo in proportion to the quantity of amyloid present and thereby enables diagnosis, quantification, and monitoring of amyloid to be performed scintigraphically.61 Labelled SAP uptake does not depend on ongoing amyloid deposition and the technique provides a macroscopic whole body survey that is diagnostic in most patients with systemic forms of the disease. It is most sensitive for solid viscera such as the liver, kidneys and spleen, and even though it is poor for imaging the lungs, it remains useful to determine whether amyloid is present in other organ systems. Serial SAP scintigraphy in over 1000 patients with various types of systemic amyloidosis has confirmed that amyloid deposits do regress, albeit relatively slowly, in the majority of cases when the supply of the respective fibril precursor protein is reduced (fig 5).4 ,62 ,63 This has been demonstrated systematically in patients with AA amyloidosis following intensive anti-inflammatory therapy, in patients with AL amyloidosis following chemotherapy, and in patients with familial amyloid polyneuropathy and dialysis amyloidosis following liver and renal transplantation, respectively.64

Serial SAP scintigraphs two years apart in a 66 year old man with systemic AL amyloidosis who presented with advanced renal impairment (serum creatinine 260 μmol/l). The baseline posterior whole body scintigraph (A) showed the renal amyloid deposits which regressed to almost undetectable levels following treatment with standard dose vincristine, adnamycin, and dexamethasone chemotherapy (B). The patient’s renal function recovered completely.
Electrocardiography (ECG) and echocardiography provide sensitive and quite specific information on cardiac involvement which occurs to some extent in most patients with systemic AL amyloidosis, and is a major prognostic determinant.65 ,66 The key to effective monitoring of amyloidosis and its treatment is relatively frequent (approximately six monthly) repetition of these investigations, bearing in mind that organ function may not closely reflect amyloid load. A scheme for managing the patient with respiratory amyloidosis is shown in fig 6.

Scheme for management of patients with respiratory tract amyloidosis.
Management
Thorough evaluation of respiratory tract amyloidosis is required to determine the type and need for treatment. The paucity of controlled clinical trials means that management decisions have to be made on an individual empirical basis. Broadly, systemic chemotherapy is indicated for systemic AL amyloidosis67 and local intervention for its localised forms.
Symptomatic laryngeal amyloidosis is usually amenable to endoscopic surgery.68 ,69 Larger masses may be removed by laryngofissure but staged endoscopic excisions are probably as effective and cause less morbidity.70 ,71 Carbon dioxide laser evaporation may be particularly useful for small recurring lesions since it is minimally invasive and thereby helps to preserve laryngeal function.72 ,73 Local and systemic corticosteroids have no effect on laryngeal amyloidosis.74
The management of tracheobronchial amyloidosis is also largely dependent upon symptoms and may involve intermittent bronchoscopic resection, surgical resection, carbon dioxide laser ablation, and Nd:YAG laser therapy.75 ,76 Repeated bronchoscopic intervention is thought to be preferable and safer than open surgery.28 Stenting may have a role for narrowing of the airways and complications thereof.
Once the diagnosis is clear, nodular parenchymal amyloidosis rarely requires intervention.28 ,77 In contrast, diffuse parenchymal amyloidosis is usually a systemic phenomenon with a poor prognosis.29 Treatment with corticosteroids or irradiation do not influence its course48 and, assuming that it is of AL type, chemotherapy to suppress the underlying plasma cell dyscrasia should be considered. Low dose oral melphalan and prednisolone produces a clinical response in only 20–30% of patients, whereas more intensive (but less well studied) infusional chemotherapy regimens and peripheral stem cell transplantation are more promising (figs 5 and7).67 ,78 ,79

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Chest radiograph and (B) thoracic CT scan of a patient with systemic AL amyloidosis who presented with cough and dyspnoea due to diffuse pulmonary parenchymal involvement. She underwent treatment with high dose melphalan and autologous stem cell rescue with subsequent improvement in symptoms and respiratory function. She remains completely well 19 months after treatment.
Recognition that amyloid deposition may have a vital role in Alzheimer’s disease has led to new vigour in the search for novel anti-amyloid drugs, some of which have already been tested in vivo. Agents are presently being sought that could stabilise amyloid precursor proteins in their normal conformation, inhibit fibril formation or propagation, or enhance the fibril degradation that evidently does occur in most patients. Candidate drugs under development include highly charged polysulphonated GAG mimetics and SAP binding inhibitors. It has been confirmed that SAP contributes to amyloidogenesis in vivo,18 and that small molecules which compete with the naturally occurring GAGs in their binding to amyloid fibrils substantially inhibit amyloid deposition.19 ,80Sophisticated assays that can distinguish amyloid fibril formation and propagation have been reported and will hopefully enable generic fibrillogenesis inhibitors to be identified within the near future.
There is an online support and discussion group for patients with amyloidosis and their relatives which is also subscribed to by many treating physicians; the address is http://www.acor.org/amyloid.html. In addition, the International Myeloma Foundation (IMF) has an increasing interest in amyloidosis.
Conclusions and prospects for the future
The manifestations, clinical significance, and prognosis of respiratory tract amyloidosis vary substantially depending on its aetiology and anatomical distribution. Each patient therefore requires thorough evaluation to determine their optimal management. Although treatment is presently restricted to supportive and local measures, along with endeavours to suppress the supply of fibril precursors, this approach is frequently very successful.
However, new approaches to treatment are required and agents that could stabilise amyloid precursor proteins in their normal conformation, inhibit fibril formation or propagation, and/or enhance fibril degradation are currently being sought. Such “anti-amyloid” drugs, some of which are presently under development, should be available for treatment of patients with amyloidosis in the near future.
Acknowledgments
This work was supported in part by Medical Research Council Programme grant G7900510 to MBP and PNH. We thank Dr C Cousins, Consultant Radiologist, Hammersmith Hospital for assistance with the radiology.