Article Text

Statistics from Altmetric.com
Definition and aetiologies
The small vessel vasculitides of the lungs1-3 (table1) are inflammatory destructive processes that affect the arterioles, venules, and alveolar capillaries located within the interstitial compartment. An intense infiltration of activated neutrophils results in fibrinoid necrosis and dissolution of arteriolar and venular walls, thus compromising the vascular lumen. Accompanying the arteriolitis and venulitis is a distinct interstitial (alveolar wall) component referred to as necrotising pulmonary capillaritis.4-9 Necrotising pulmonary capillaritis can occur in isolation, in the absence of histological evidence of arteriolitis or venulitis. It is recognised by a marked interstitial neutrophilic infiltration and many of these cells are undergoing leucocytoclasis or fragmentation (fig 1). Because these neutrophils are constantly undergoing cell death (apoptosis), pyknotic cells and nuclear fragments (dust) accumulate within the lung parenchyma. The interstitial space becomes broadened by oedema, fibrin, and the neutrophilic infiltrate and eventually undergoes fibrinoid necrosis (fig 2). During this process the integrity of interstitial capillaries is damaged, permitting red blood cells to traverse the now incompetent alveolar capillary basement membranes and freely enter the interstitial compartment and flood alveolar spaces. Clinically this is referred to as diffuse alveolar haemorrhage, and this accompanies most episodes of small vessel vasculitis in the lungs regardless of aetiology. Fibrin and neutrophils also traverse the damaged alveolar capillary basement membranes and enter the alveolar spaces with the red blood cells. Other histological features in the small vessel vasculitides of the lung include arteriolar and capillary thrombosis, organising haemorrhage, epithelial type 2 cell hyperplasia, and eventually the accumulation of free parenchymal haemosiderin and haemosiderin containing macrophages in alveolar spaces (fig 3). With time, and after repeated episodes of diffuse alveolar haemorrhage due to pulmonary capillaritis, both interstitial pulmonary fibrosis and a progressive obstructive lung disease with the physiological and computed tomographic appearance of emphysema have been described.9 ,10
Aetiology of small vessel vasculitis of the lungs (pulmonary capillaritis) in order of relative frequency1–3
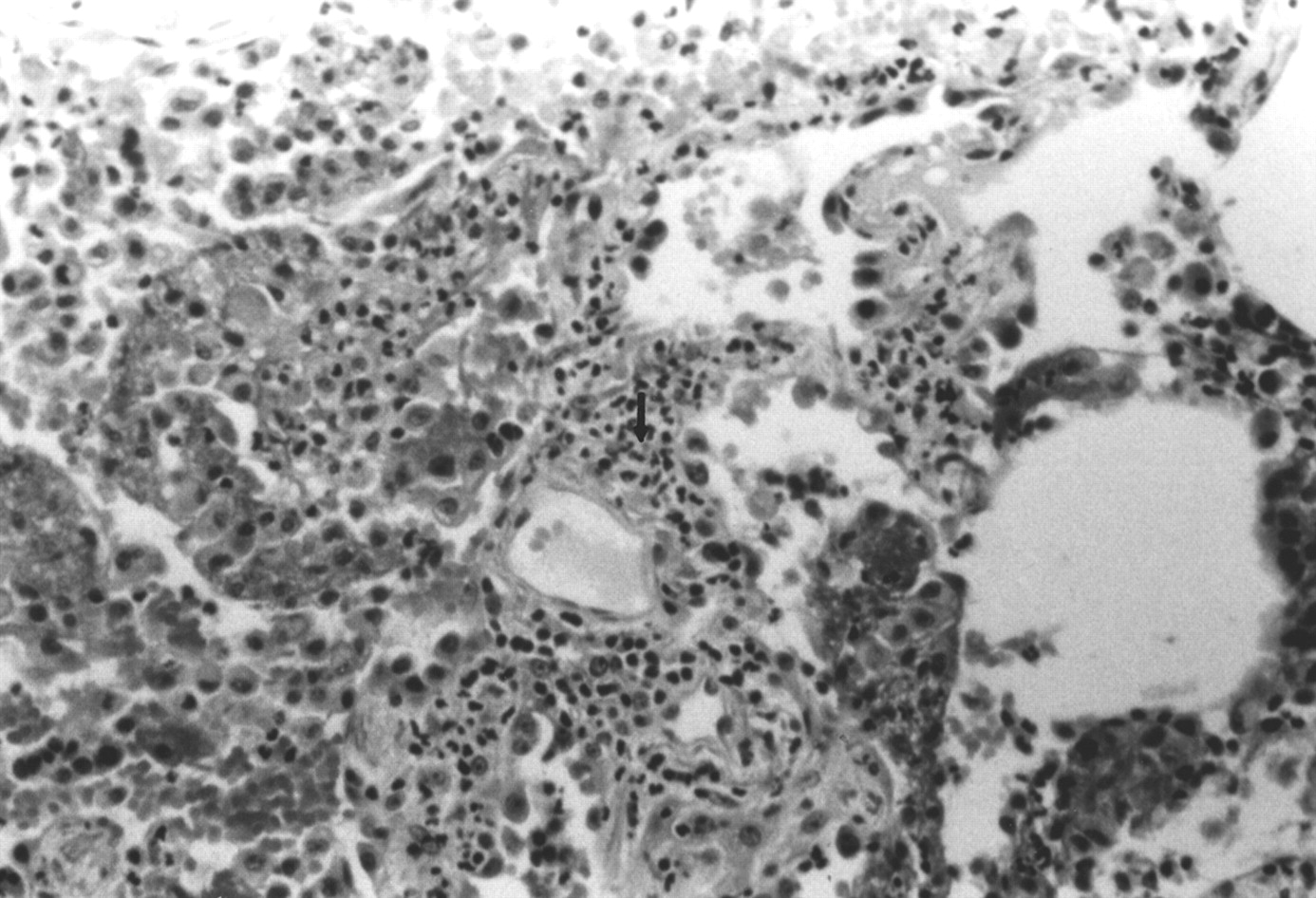
Necrotising pulmonary capillaritis in isolated pauci-immune pulmonary capillaritis. The interstitial space is expanded by infiltrating neutrophils (arrow), many of which appear fragmented.
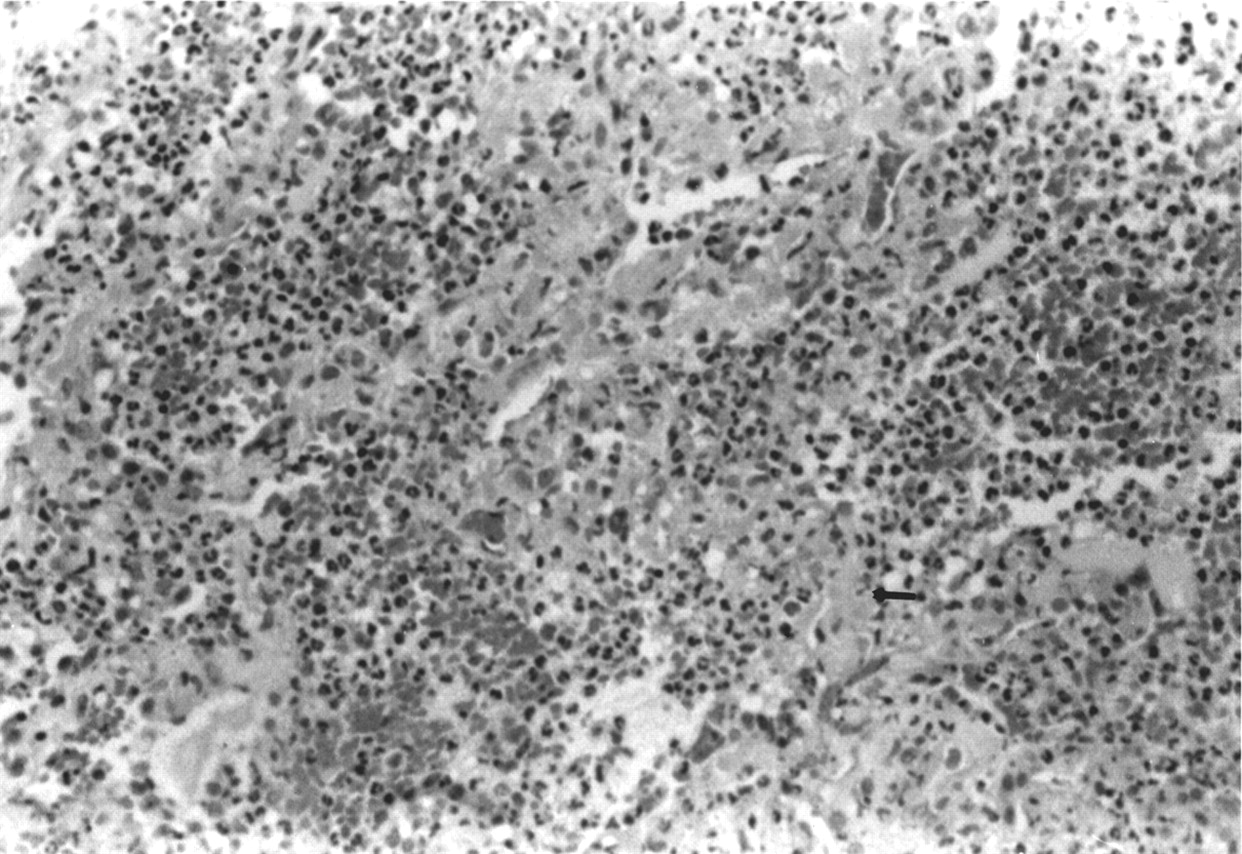
Necrotising pulmonary capillaritis in Wegener's granulomatosis. Note the destruction and fibrinoid necrosis of the interstitium (arrow). There is a striking neutrophilic infiltration, as well as diffuse alveolar haemorrhage. The patient did not survive.

Organising diffuse alveolar haemorrhage secondary to necrotising pulmonary capillaritis in a patient with microscopic polyangiitis. In addition to the fibroblastic proliferation and new collagen formation, there is residual diffuse alveolar haemorrhage and capillaritis. The patient recovered.
Small vessel vasculitis of the lungs occurs in the setting of systemic vasculitis, collagen vascular disease, or another systemic illness associated with the production of autoantibodies, as outlined in table1. It can also be isolated to the lungs and, in this case, it is referred to as pauci-immune isolated pulmonary capillaritis. The isolated form of small vessel vasculitis of the lungs is one of the most frequent causes of pulmonary capillaritis and diffuse alveolar haemorrhage.11-14
Clinical manifestations and diagnosis
Haemoptysis is the most common clinical manifestation of small vessel vasculitis involving the lungs, reflecting the underlying diffuse alveolar haemorrhage. However, up to 33% of patients suffering from any of the conditions listed in table 1 may have significant intra-alveolar bleeding without this symptom.1 ,4 If this is the case, a chest radiograph indicating new unexplained bilateral alveolar infiltrates in the face of a falling haemoglobin and a haemorrhagic sequential bronchoalveolar lavage (BAL), in which sequential aliquots appear more bloody, points to the potential underlying histology. Most patients with diffuse alveolar haemorrhage are too ill to undergo this procedure, however, but measurements of the carbon monoxide transfer coefficient corrected for lung volume (Kco) may show sequential increases over time because of the heightened availability of intra-alveolar haemoglobin for combination with carbon monoxide. It should be understood, however, that there are other underlying pulmonary disorders which can result in diffuse alveolar haemorrhage. These include diffuse alveolar damage from a number of causes, pulmonary veno-occlusive disease, and bland non-inflammatory pulmonary haemorrhage, which can occur with mitral stenosis, anticoagulation and thrombolytic therapy, various coagulopathies, idiopathic pulmonary haemosiderosis, and some cases of Goodpasture's syndrome.1-3
Other symptoms of diffuse alveolar haemorrhage include dyspnoea, cough, and a low grade fever suggesting an infectious pneumonia. These symptoms usually evolve over several days to a week, although more subacute cases with intermittent haemoptysis can also occur. All age groups are affected and recurrent episodes of diffuse alveolar haemorrhage following successful treatment, particularly in patients with systemic vasculitis or a collagen vascular disease, are to be expected. In our experience, up to 50% of episodes of diffuse alveolar haemorrhage result in respiratory failure severe enough to require assisted ventilation.
The medical history, including both prescribed and illicit drug use, is important in a patient who presents with isolated diffuse alveolar haemorrhage. Crack cocaine use is a frequent cause of diffuse alveolar haemorrhage, resulting from diffuse alveolar damage. Prescribed drugs such as diphenylhydantoin and propylthiouracil have been reported to cause hypersensitivity vasculitis and a systemic small vessel vasculitis which may include the lungs.15 Diffuse alveolar haemorrhage and pulmonary capillaritis can occur following transretinoic acid therapy for promyelocytic leukaemia.16Bland pulmonary haemorrhage with normal alveolar histology can also result from anticoagulant use, thrombolytic therapy, and thrombocytopenia.
If diffuse alveolar haemorrhage develops in a patient with a previously defined systemic vasculitis or collagen vascular disease, the diagnosis is relatively easy to establish; however, it is not unusual for diffuse alveolar haemorrhage with underlying necrotising pulmonary capillaritis to represent the initial clinical event of a systemic disease or, as previously stated, the syndrome can be confined to the lungs. Clinical abnormalities suggesting systemic involvement should be looked for.17-19 These include sinusitis, dermatological leucocytoclastic vasculitis, iridiocyclitis, synovitis, and glomerulonephritis. A urinalysis demonstrating proteinuria, haematuria, and red blood cell casts suggests the possibility of an underlying focal segmental necrotising glomerulonephritis, a rapidly progressive glomerulonephritis common to the systemic vasculitides, systemic lupus erythematosus (SLE), Goodpasture's syndrome, and pauci-immune idiopathic glomerulonephritis (fig 4).
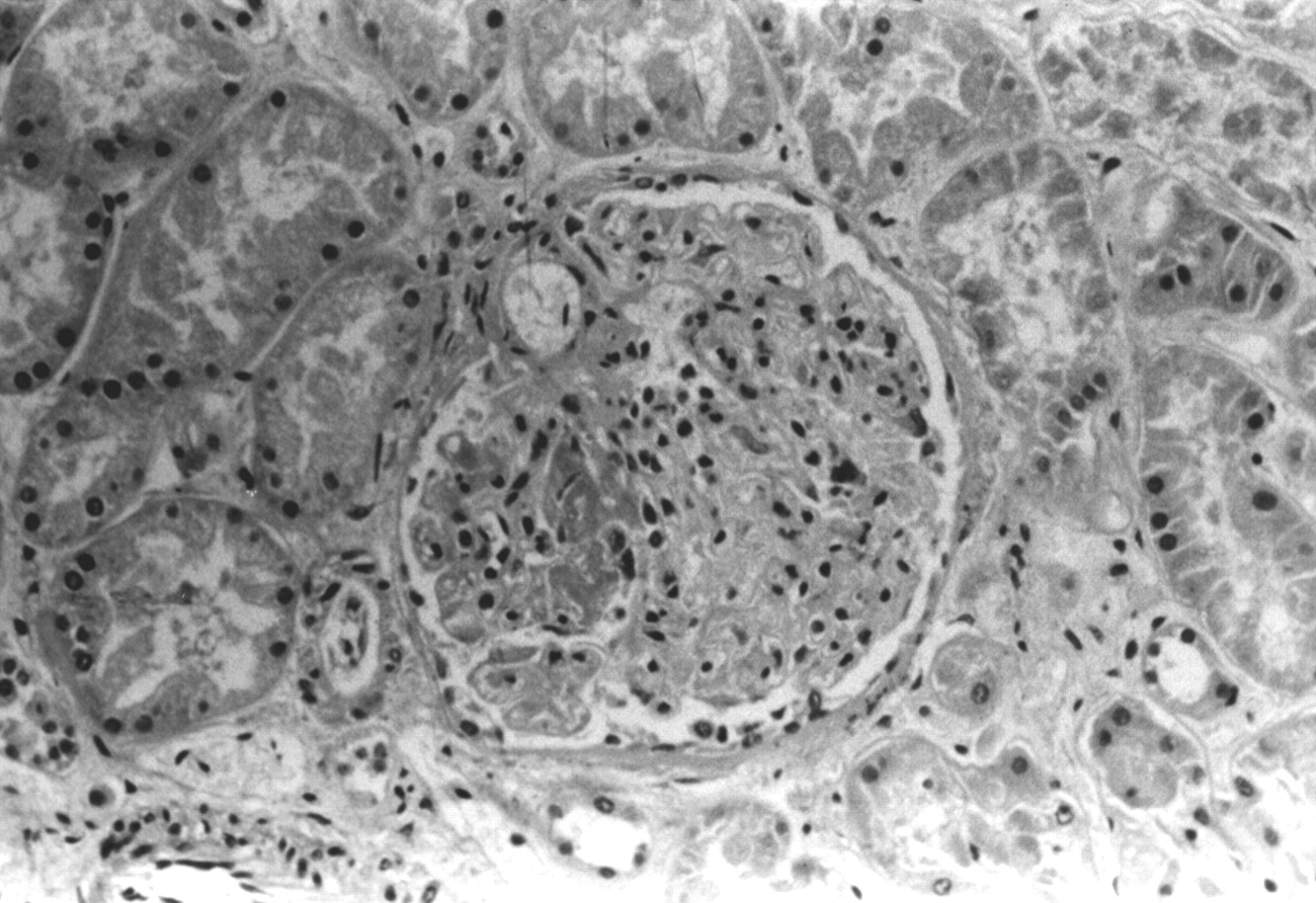
Focal segmental necrotising glomerulonephritis in a patient with SLE. This is an advanced case with glomerular crescent formation.
The chest radiograph is non-specific and indicates air space disease that can be either patchy or diffuse in appearance (fig 5). The presence of Kerley B lines on the chest radiograph suggests either another cause of diffuse alveolar haemorrhage such as pulmonary veno-occlusive disease, mitral stenosis, or possibly pulmonary oedema due to myocarditis which can potentially complicate a systemic vasculitis or collagen vascular disease. A computed tomographic lung scan offers little advantage over the chest radiograph in determining the aetiology of diffuse alveolar haemorrhage.

Chest radiograph showing non-specific diffuse patchy air space disease in a patient with isolated pauci-immune pulmonary capillaritis.
Non-specific increases in the white blood cell and platelet counts are common. Thrombocytopenia, on the other hand, may indicate idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, disseminated intravascular coagulation, or leukaemia, all conditions associated with bland pulmonary haemorrhage.20-22 Thrombocytopenia could also be a clue to the diagnosis of the primary antiphospholipid syndrome or SLE, both associated with antiphospholipid antibodies and underlying pulmonary capillaritis and diffuse alveolar haemorrhage.11 ,23 ,24
Goodpasture's syndrome, a condition associated with either bland pulmonary haemorrhage or pulmonary capillaritis, is confirmed by the presence of antibasement membrane antibody in the serum or continuous linear deposits of IgG and complement along the basement membranes in renal or lung tissue (fig 6).25 ,26 Some 5–10% of patients with Goodpasture's syndrome present without clinical evidence of active renal disease.27-33 In this case, serum antibasement membrane antibody can be absent and the diagnosis can only be established by demonstrating linear immunofluorescence in lung or renal tissue. In 20% of patients with diffuse alveolar haemorrhage secondary to SLE, the diffuse alveolar haemorrhage episode represents the first clinical manifestation of the systemic disease.34-38 In this instance the demonstration of serum antinuclear antibodies and antibodies directed against double stranded desoxyribonucleic acid (DNA), as well as a reduced serum complement, helps establish the diagnosis. If lung or renal tissue is available, immune complexes consisting of IgG and complement deposited in a granular fashion are visible along alveolar walls, around small blood vessels, and within glomerular capillaries.35 ,39-43Alternatively, if lung or renal immunofluorescence reveals IgA immune complexes, the diagnosis of either Henoch-Schoenlein purpura or IgA nephropathy, two less frequent causes of diffuse alveolar haemorrhage and pulmonary capillaritis, should be considered.41 ,43-47 In patients with Wegener's granulomatosis, microscopic polyangiitis, isolated pauci-immune pulmonary capillaritis, or idiopathic pauci-immune glomerulonephritis, lung immunofluorescence is negative or pauci-immune (no complement or immunoglobulin deposition).

An immunofluorescent stain of a lung biopsy specimen from a patient with Goodpasture's syndrome demonstrating linear deposition of IgG.
Serum antiphospholipid antibodies should be measured to exclude the primary antiphospholipid syndrome, especially in patients with a history of thrombophlebitis and thrombocytopenia.23 ,24 The detection of antineutrophil cytoplasmic antibodies (ANCA) in the serum is often helpful. p-ANCA (antibody to cytoplasmic myeloperoxidase) positivity is seen in microscopic polyangiitis, some cases of isolated pulmonary capillaritis, idiopathic pauci-immune glomerulonephritis, propylthiouracil-induced systemic vasculitis, and the unusual case of diffuse alveolar haemorrhage due to Churg-Strauss syndrome.12 ,13 ,18 ,48-58 In the latter condition the presence of peripheral eosinophilia and increased serum IgE levels support the diagnosis in the setting of diffuse alveolar haemorrhage. Serum c-ANCA (antibodies to cytoplasmic proteinase 3) positivity in the setting of diffuse alveolar haemorrhage, on the other hand, is virtually diagnostic of Wegener's granulomatosis.59
The diagnosis of diffuse alveolar haemorrhage is usually not difficult to establish even in patients without haemoptysis. A low or falling haematocrit and sequential BAL fluid samples which are persistently haemorrhagic secure the diagnosis in patients with an acute pulmonary syndrome and pneumonic-type radiographic infiltrates. The specific diagnosis of small vessel vasculitis including pulmonary capillaritis requires lung tissue for confirmation, particularly in those patients who present for the first time with unexplained diffuse alveolar haemorrhage and who do not have an established diagnosis of an underlying systemic disease. A thoracoscopic biopsy is recommended for patients with diffuse alveolar haemorrhage who do not have a prior diagnosis of a systemic disease at presentation or who have unexplained isolated diffuse alveolar haemorrhage and in whom serological studies do not support a specific diagnosis. There is no evidence to suggest that diffuse alveolar haemorrhage increases operative morbidity or mortality.
Pauci-immune pulmonary capillaritis can be associated with or without serum p-ANCA positivity. Isolated diffuse alveolar haemorrhage also appears in the lung limited forms of Goodpasture's syndrome, the primary antiphospholipid syndrome, and occasionally in the collagen vascular diseases. On the other hand, the finding of bland pulmonary haemorrhage at biopsy in a patient with lung limited disease could represent idiopathic pulmonary haemosiderosis. In a patient with unexplained bland pulmonary haemorrhage, echocardiography is indicated to exclude the possibility of previously undetected mitral stenosis.
Immunopathogenesis
In small vessel vasculitis of the lungs it is likely that the infiltration and subsequent activation of neutrophils leads to the release of cytoplasmic proteolytic enzymes and oxidants, which are responsible for necrosis of the interstitial matrix and vascular walls and the loss of integrity of the alveolar capillary basement membranes. In biopsy specimens of less involved areas of the lung from patients with small vessel vasculitis, neutrophils can be seen accumulating in an intact interstitial space (fig 7). Although the chemotactic stimulus remains unknown, there is evidence to suggest that the interstitial neutrophils undergo an oxidative burst and degranulate, resulting in the destruction of the alveolar wall.60-62 However, the stimuli which attract, prime, and activate the neutrophils in vivo are not known.

Early pulmonary capillaritis in an area of the lung biopsy without overt diffuse alveolar haemorrhage. Note the infiltration of alveolar walls by neutrophils, but without oedema or necrosis.
Although there is a potential role for ANCA in the pathogenesis of the vasculitis in Wegener's granulomatosis and microscopic polyangiitis, some patients with these conditions do not have antibodies. Furthermore, small vessel vasculitis in conditions such as isolated pauci-immune pulmonary capillaritis, most cases of diffuse alveolar haemorrhage in collagen vascular disease and lung allograft rejection are ANCA negative.11 ,34 ,60-64 Nonetheless, there is evidence both in vitro and in vivo to suggest a potential role for ANCA in the pathogenesis of ANCA positive vasculitis.57 ,59 ,65-68 It has been postulated that, either via stimulation by bacterial products such as lipopolysaccharide or formyl tripeptides, or by cytokines such as interleukin 1 or transforming growth factor β, circulating neutrophils become primed and express either intracytoplasmic proteinase 3 or myeloperoxidase on their cell surfaces. Circulating ANCA then attach to the primed neutrophil surfaces causing destruction of the neutrophilic cell membrane resulting in the subsequent release of intracytoplasmic toxic products with resultant matrix and endothelial injury.57 ,65 It has also been shown that the endothelial cell contributes to this injury by increasing its expression of adhesion molecules which enhance the attachment of primed neutrophils to endothelial surfaces.11 ANCA have also been shown to have direct cytotoxic effects on endothelial cells.68-70A mouse model of systemic vasculitis has been developed which demonstrates granulomatous lung lesions following the injection of human c-ANCA.71 ,72 These animals also develop anti-idiotypic antibodies to c-ANCA, and eventually c-ANCA itself. In contrast to the pauci-immune disease in Wegener's granulomatosis, immune complexes were present. However, human c-ANCA does not recognise murine proteinase 3, evidence against the pathogenicity of ANCA in this model.73
Small vessel vasculitis of the lung may be associated with disorders of autoimmunity and tissue immune complex deposition (table 1). The formation of intravascular immune complexes via the clonal expansion of B cells in response to an antigen, possibly an infectious organism or a drug, can result in attachment to endothelial basement membranes and activation of complement. This, in turn, leads to chemotaxis and activation of inflammatory cells followed by an oxidative burst and degranulation with the release of serine proteases resulting in vascular necrosis and matrix injury. It is possible that this somewhat simplified explanation of immune complex induced small vessel vasculitis can explain the development of vasculitis in conditions such as hypersensitivity or drug induced vasculitis, SLE and possibly other collagen vascular diseases, Henoch-Schoenlein purpura, cryoglobulinaemic vasculitis, and the primary antiphospholipid syndrome with pulmonary capillaritis.18 ,23 ,36 ,42 ,74-77
Selected small vessel vasculitides of the lung
ISOLATED PAUCI-IMMUNE PULMONARY CAPILLARITIS
Isolated pauci-immune pulmonary capillaritis causing diffuse alveolar haemorrhage is a recently described syndrome that occurs with and without serum p-ANCA positivity.11-14 In a series of 29 biopsy proven cases of pulmonary capillaritis from all causes, isolated pauci-immune pulmonary capillaritis without serum ANCA positivity was the most frequent diagnosis, being found in eight cases. In patients with isolated small vessel vasculitis of the lungs no clinical or serological evidence was present either initially or during a prolonged follow up period (mean 43 months) to suggest the development of a systemic disease.11 Six of the eight patients had respiratory failure at presentation, four requiring assisted ventilation. Following treatment with corticosteroids and cyclophosphamide, seven survived the initial event and recovered. Two of the seven had recurrent exacerbations of diffuse alveolar haemorrhage and one subsequently died of respiratory failure. A necropsy showed that the disease was confined to the lungs. There are also several reports of p-ANCA positive isolated pauci-immune pulmonary capillaritis,12 ,13 though follow up data are not available to determine whether a systemic disease developed. It is possible that p-ANCA positive pauci-immune pulmonary capillaritis is similar to pauci-immune idiopathic glomerulonephritis, which is also p-ANCA positive and represents an isolated renal small vessel vasculitis.78-80 Patients with this isolated form of renal vasculitis occasionally develop a systemic disease with diffuse alveolar haemorrhage. These cases are difficult to differentiate from microscopic polyangiitis.
It is also the opinion of the authors that there are cases of isolated diffuse alveolar haemorrhage in adults previously ascribed to idiopathic pulmonary haemosiderosis, which actually represent isolated pauci-immune pulmonary capillaritis.
WEGENER'S GRANULOMATOSIS
Diffuse alveolar haemorrhage secondary to a small vessel vasculitis may be the initial and only pulmonary manifestation of Wegener's granulomatosis.78-92 Approximately 40 cases of diffuse alveolar haemorrhage secondary to Wegener's granulomatosis with capillaritis, arteriolitis, and venulitis representing the sole histological manifestations in the lung have been reported. Evidence for renal involvement with an underlying focal segmental necrotising glomerulonephritis is usually present and, in this case, if the serum c-ANCA is positive, the diagnosis of Wegener's granulomatosis is established. Although the therapeutic implications remain the same, there are cases of small vessel vasculitis of the lung eventually diagnosed as Wegener's granulomatosis in which the serum p-ANCA is positive, making this diagnosis initially indistinguishable from microscopic polyangiitis.93-100 The diagnosis is eventually established when more typical clinical and histological features of Wegener's granulomatosis evolve. This may take several years.25 The early mortality is considerably higher in patients with Wegener's granulomatosis who present with diffuse alveolar haemorrhage and pulmonary capillaritis than in those with more typical granulomatous vasculitic lung lesions.25 ,82-85 ,101 The cause of death is usually renal or respiratory failure or sometimes a complicating infection secondary to the intense immunosuppressive therapy.
A small vessel vasculitis of the lung often coexists with the more typical necrotising granulomatous vasculitis of Wegener's granulomatosis. This was seen in 31% of patients with Wegener's granulomatosis who underwent open lung biopsy.85 Diffuse alveolar haemorrhage may also present in Wegener's granulomatosis during an exacerbation in an established case.90 ,102 ,103
MICROSCOPIC POLYANGIITIS
Microscopic polyangiitis is considered to be the small vessel variant of polyarteritis nodosa. In addition to differences in the size of the vessels involved, other distinguishing features from polyarteritis nodosa include the following: microscopic polyangiitis is almost never associated with a prior hepatitis B or C infection; lung involvement is rare in polyarteritis nodosa while a small vessel vasculitis causing diffuse alveolar haemorrhage is reported to occur in 10–30% of patients with microscopic polyangiitis6 ,7 ,9 ,12 ,104-113; the rapidly progressive glomerulonephritis that occurs in 80–100% of patients with microscopic polyangiitis does not occur in polyarteritis nodosa.114 ,115 Other systemic involvement is similar and includes arthritis, myositis, gastrointestinal bleeding secondary to mucosal vasculitis, peripheral neuropathy, and sinusitis. Both conditions can be p-ANCA positive.109 ,115 Some cases of pauci-immune idiopathic glomerulonephritis complicated by diffuse alveolar haemorrhage and localised forms of pulmonary capillaritis are also p-ANCA positive.12 ,13 ,78-80
The presence of a small vessel vasculitis involving the lungs increases the early mortality in microscopic polyangiitis.8 ,17 ,25 ,87 ,106-109 ,116 ,117 Up to 25% of patients will die during the first episode of diffuse alveolar haemorrhage. In those who survive the response to treatment is good but recurrent episodes are to be expected, as with other types of vasculitis. With recurrent episodes of diffuse alveolar haemorrhage, both obstructive lung disease (thought to be emphysema) and pulmonary fibrosis have been described as chronic complications (fig8).9-11 ,118

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Chest radiograph of a patient with diffuse alveolar haemorrhage secondary to microscopic polyangiitis. (B) Nine years later, following multiple recurrences of diffuse alveolar haemorrhage secondary to biopsy proven necrotising pulmonary capillaritis, there is hyperinflation. Physiological tests indicated moderately severe obstructive lung disease.
COLLAGEN VASCULAR DISEASE
In a recent review of the complications of SLE, diffuse alveolar haemorrhage occurred in 15 patients (4%). In 12 patients the diffuse alveolar haemorrhage occurred in an established case (30–35 months following the initial diagnosis), but in 23% it was the presenting manifestation of SLE.34 All but one patient had an active glomerulonephritis. The early mortality was 50%, the highest mortality for any known cause of diffuse alveolar haemorrhage. The lung histology of diffuse alveolar haemorrhage in SLE was previously reported as bland pulmonary haemorrhage or diffuse alveolar damage.12 ,35 ,39-41 119-128 More recent studies indicate that a small vessel vasculitis in the form of arteriolitis, venulitis, and capillaritis was the most frequent association with diffuse alveolar haemorrhage in SLE.34-36 ,74 Immune complex deposition is often found in the lungs of patients with SLE small vessel vasculitis and diffuse alveolar haemorrhage.1 ,34-36 The presence of immune deposits in a granular distribution distinguishes the diffuse alveolar haemorrhage of SLE from that of Wegener's granulomatosis, microscopic polyangiitis (pauci-immune), and Goodpasture's syndrome (linear deposition).
A small vessel vasculitis (pulmonary capillaritis) was recently reported in several patients with polymyositis.61 In these cases the lung and muscle disease appeared simultaneously and in one patient serum anti Jo-1 antibodies were present. Serum ANCA measurements were negative. There was a good response to standard vasculitis therapy (see below). Both rheumatoid arthritis and mixed connective disease can be complicated by diffuse alveolar haemorrhage and pulmonary capillaritis and it almost always appears in established collagen vascular disease.62 In these conditions diffuse alveolar haemorrhage can present either as a small vessel vasculitis localised to the lung or as a component of a more generalised vasculitis which often includes glomerulonephritis. Serum p-ANCA levels may be positive.62 ,129-132 Pulmonary capillaritis can complicate the course of scleroderma133-135 and the primary antiphospholipid syndrome.23 ,24 ,136 In the latter there is usually a history of prior thrombotic events and thrombocytopenia is present.
Treatment
The treatment of small vessel vasculitis of the lung is largely the same, regardless of aetiology or whether it is isolated to the lung or a component of a systemic disease. Corticosteroids (prednisone) and cyclophosphamide are the mainstays of treatment.137-140Prednisone is given orally in a dose of 1 mg/kg/day or intravenously as methylprednisolone in a dose of 250–1000 mg/day for 3–5 days. Following intravenous administration, oral treatment is initiated as outlined above. Following a therapeutic response, corticosteroids are tapered over the ensuing 2–6 months. In addition, cyclophosphamide is given orally in a dose of 2 mg/kg/day and continued for 6–12 months, then tapered over the next several months. For patients who require assisted ventilation, intravenous cyclophosphamide is initially given as a single dose (1 g/m2) and oral therapy is then started after 2–4 weeks. With this regimen up to 20% of patients may develop Pneumocystis carinii pneumonia so prophylaxis with trimethoprim (160 mg)-sulfamethoxazole (800 mg) three days a week is recommended.141-143 Prolonged cyclophosphamide therapy has a high incidence of side effects including infection, haemorrhagic cystitis, transitional cell carcinoma of the bladder, and myelodysplasia.138 MESNA may have a role in the prevention and treatment of the haemorrhagic cystitis. The incidence of bladder cancer in one study of 145 patients with Wegener's granulomatosis treated with cyclophosphamide was 5% at 10 years and 16% at 15 years.144 Persistent non-glomerular haematuria identified a subgroup with an increased risk of this complication. Other risk factors included a cumulative dose of cyclophosphamide which exceeded 100 g and total duration of treatment of more than 2.7 years. Recurrences of diffuse alveolar haemorrhage are not unusual as treatment is being tapered or even after long periods of stability off treatment.
Alternative immunosuppressive drugs for patients with vasculitis who are unable to tolerate cyclophosphamide include azathioprine and methotrexate. There are, however, only limited data to support their long term efficacy.145 ,146 In patients unresponsive to standard cytotoxic and immunosuppressive therapy, plasmapheresis has been recommended.147 ,148 Unlike Goodpasture's syndrome where plasmapheresis is of definite clinical benefit, particularly in patients with uncontrolled diffuse alveolar haemorrhage, the results in systemic vasculitis are mixed and further trials are required.146-155 Pooled intravenous gamma globulin has been attempted, usually in patients with typical Wegener's granulomatosis. The results are contradictory, possibly due to the variability of the effectiveness of the pooled samples.156-160 Other treatments for Wegener's granulomatosis refractory to or intolerant of standard therapy have included humanised monoclonal antibodies specific to lymphocyte subsets.161 Remission was obtained in all six patients in one report; however, the disease recurred after withdrawal of treatment, but responded promptly to reinstitution of treatment.
Small vessel vasculitis of the lung and subsequent diffuse alveolar haemorrhage can be a catastrophic illness if recognition and treatment are delayed. Diagnosis is often aided by other systemic findings, associated illness, and serological studies. Patients with unexplained isolated diffuse alveolar haemorrhage should undergo a lung biopsy with immunofluorescent studies and routine histological tests. Effective treatment is available but patients require close monitoring because of potential complications of treatment and the tendency for all causes of small vessel vasculitis to recur.
Acknowledgments
Supported by the National Heart, Lung and Blood Institute Specialized Center of Research Grant HL-27353-04.