



Why is a paediatric respiratory specialist integral to the paediatric rheumatology clinic?
- 1Dept of Paediatric Respiratory Medicine, Nottingham Children's Hospital, Nottingham University Hospitals, Nottingham, UK
- 2Dept of Radiology, Nottingham University Hospitals, Nottingham, UK
- Correspondence: E-mail: jayesh.bhatt{at}nuh.nhs.uk
Abstract
Systemic connective tissue diseases (CTDs) are characterised by the presence of autoantibodies and multiorgan involvement. Although CTDs are rare in children, they are associated with pulmonary complications, which have a high morbidity and mortality rate. The exact pathophysiology remains unclear. The pleuropulmonary complications in CTD are diverse in their manifestations and are often complex to diagnose and manage.
The most common CTDs are discussed. These include juvenile systemic lupus erythematosus, juvenile dermatomyositis, juvenile systemic sclerosis, Sjögren's syndrome and mixed connective tissue disease. We describe the clinical features of the pleuropulmonary complications, focusing on their screening, diagnosis and monitoring. Treatment strategies are also discussed, highlighting the factors and interventions that influence the outcome of lung disease in CTD and pulmonary complications of treatment.
Early detection and prompt treatment in a multidisciplinary team setting, including respiratory and rheumatology paediatricians and radiologists, is paramount in achieving the best possible outcomes for these patients.
Abstract
Pleuropulmonary complications of CTD, though rare in paediatrics, can be associated with high morbidity and mortality. Joint management by respiratory and rheumatology paediatricians is recommended. Treatment includes steroids and other immunomodulators. https://bit.ly/2MCvEpp
Educational aims
To discuss the pulmonary manifestations in children with CTD.
To outline the diagnostic modalities used to diagnose and monitor pulmonary manifestations in CTD.
To review the various treatment strategies in CTD-related lung disease and the importance of a multidisciplinary approach to management.
Connective tissue diseases (CTD) encompass a group of heterogeneous systemic inflammatory diseases characterised by the presence of circulating autoantibodies and autoimmune-mediated multiorgan system involvement [1]. These diseases are rare in children [2]. Amongst the most common are juvenile systemic lupus erythematosus (JSLE), juvenile dermatomyositis (JDM), juvenile systemic sclerosis (JSSc) and mixed connective tissue disease (MCTD).
Other conditions encountered in the paediatric rheumatology service can also be associated with pulmonary manifestations, for instance systemic vasculitis and autoinflammatory disorders such as systemic juvenile idiopathic arthritis.
Clinical presentations of autoimmune CTD are extremely variable and can be associated with many systemic complications including pulmonary complications [1]. It is difficult to assess the true prevalence of pulmonary complications in CTD as they are often influenced by the acuity, chronicity and complexity of the causative CTD. The entire pulmonary system is vulnerable to injury, and interstitial lung disease (ILD) is one of the sinister complications.
Many children are initially asymptomatic from a pulmonary perspective, and unless clinicians are vigilant with a high degree of clinical suspicion, these complications may go undetected. This will enable early diagnosis and management where pulmonary involvement occurs in the context of a known diagnosis of CTD. Very occasionally, a diagnosis of CTD may come to light when a child/young adult with diffuse lung disease is being investigated as pulmonary involvement may be the first or only manifestation of CTD.
Both respiratory and rheumatology paediatricians need a good understanding of how to monitor, investigate and manage these diseases appropriately, because early therapy may help prevent serious and permanent sequelae, thus improving patient outcomes [3]. This is best achieved with joint specialist paediatric rheumatology and respiratory reviews in a multidisciplinary setting [3]. In addition, clinicians need to be aware of pulmonary complications of the immunomodulatory treatments, including increased susceptibility to infection and development of ILD as complications of therapy.
In this review, we discuss a variety of paediatric autoimmune CTDs, highlight their clinical presentations, potential pulmonary complications, and provide an update on treatment options.
Incidence of pleuropulmonary complications in CTD
ILD is one of the most common and clinically significant pulmonary manifestations of CTD. ILD in children is a rare condition in general, with an incidence of less than one per 100 000 [4], compared with 30 per 100 000 in adults, of which CTD-related lung disease accounts for a small proportion.
Pathophysiology
As expected in a multisystem disease, the entire pulmonary system is vulnerable to injury. Any of its compartments including chest wall, pleura, vasculature, airways and parenchyma may be independently or simultaneously affected. The pathophysiology is not completely understood and is believed to be multifactorial [5].
There is an inflammatory immune response driven by autoantibodies [6]. The lungs are exposed to external stimuli such as microorganisms and toxins, and as the lungs are highly vascular, an inflammatory process is triggered. Macrophages act as antigen presenting cells, ingesting the inhaled antigenic stimuli and causing an inappropriate activation of autoimmune CD4 T-lymphocytes and B-lymphocytes. Several distinct immune cell populations contribute to ILD [6, 7].
There is a profibrotic process which causes irreversible remodelling of the lung [8]. Stimuli like cigarette smoke, radiation, cytokines and oxidative stress can cause epithelial–mesenchymal transition. The epithelial cells lose features such as cell-to-cell adherence, gain migratory and mesenchymal properties, and eventually convert into fibroblasts and myofibroblasts producing extracellular matrix, which engenders the profibrotic repair programme [9].
In addition to these immunological and fibrotic processes, genetic factors and microbiological triggers are also believed to contribute to the pathogenesis of lung disease in CTD, particularly ILD. These changes essentially impair lung function and potentially can result in abnormal gas exchange caused by the restructuring of the interstitial spaces of the lungs [7].
The underlying disease process influences which part of the respiratory system will be predominately affected, for example the pleura shares structures and function similar to the joint synovia, and subsequently it is often the site where inflammation happens [3].
Juvenile systemic lupus erythematosus
JSLE is an autoimmune disease, driven by autoantibodies resulting in multisystem inflammation and multiorgan involvement [10]. The incidence of JSLE is 0.3–0.9 per 100 000 and it is more prevalent in non-Caucasian children [11]. It shows a female predominance after puberty, and presents in 15–20% of affected individuals before the age of 18 years [12].
JSLE is more aggressive than adult-onset disease and is linked with a higher rate of arthritis, serositis, nephritis, haematological and neurological abnormalities [13]. Within the first year of diagnosis, up to 40% have associated lung involvement, and up to 80% of paediatric patients with JSLE will experience chest complications at some point in the disease course [3]. Pleuritic chest pain is the common presenting complaint. The most common histological pattern is nonspecific interstitial pneumonia, but any pattern can be seen [14].
The pulmonary complications in JSLE can be acute or chronic (table 1).
Acute and chronic pulmonary complications of JSLE
Acute pulmonary complications in JSLE
The pleura is the most commonly affected part of the respiratory system in JSLE. Pleuritis occurs in 80% of children with JSLE [3]. In fact, serositis, which is pleuritis with effusion, is one of the diagnostic criteria for JSLE [15]. The pleural effusion can be bilateral or unilateral. Chest pain is the most common presenting respiratory symptom, whereas complaints about shortness of breath are rare [16]. A pleural rub may be heard on auscultation. Many children remain asymptomatic [17]. The prognosis for isolated pleuritis is generally good.
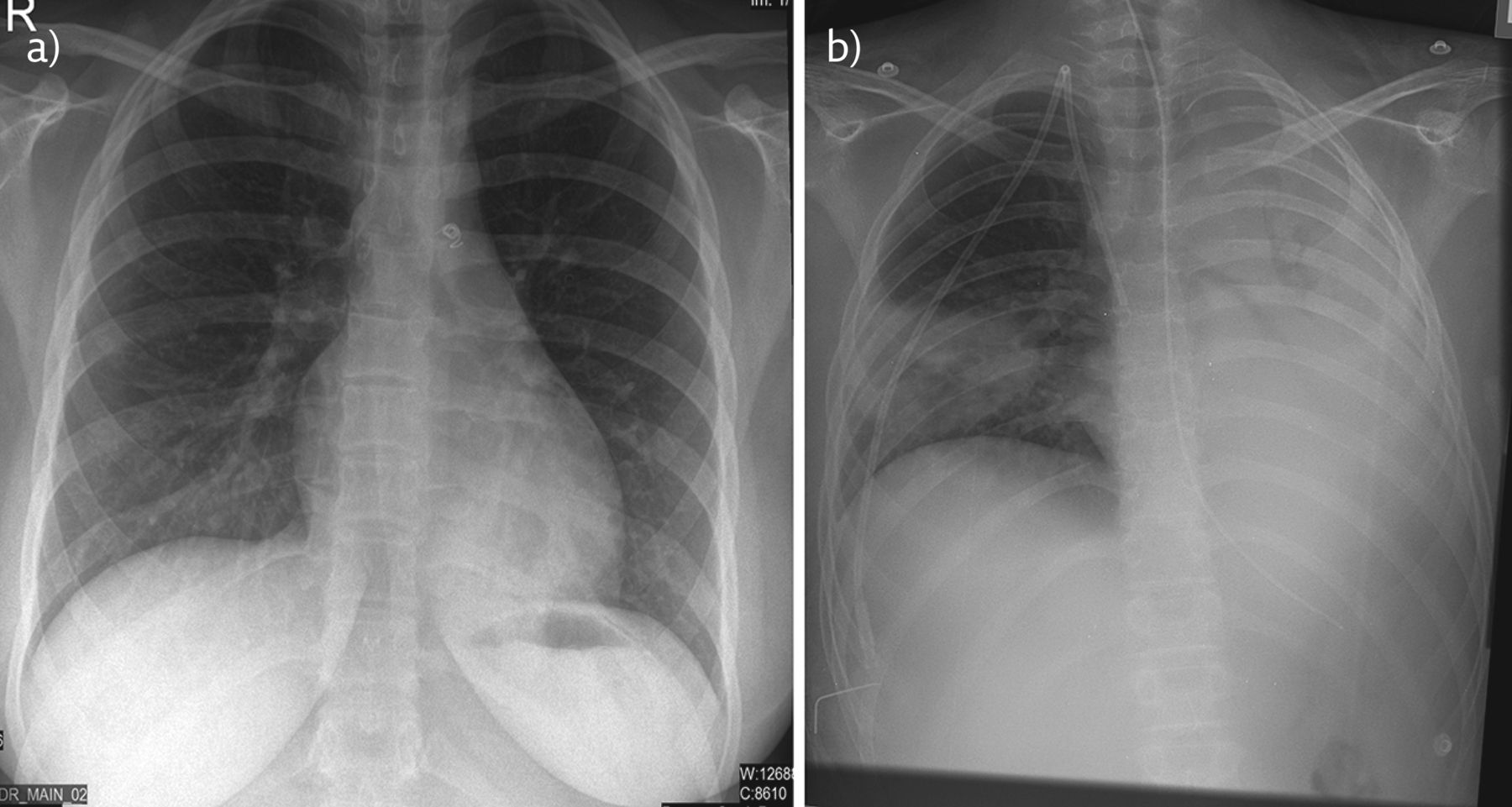
Serositis is usually diagnosed with plain chest radiography, which demonstrates pleural effusion. Examples of pleural effusion and pericardial effusions on chest imaging in a child with JSLE are shown in figure 1.

a) Chest radiograph of child with JSLE: the PA radiograph demonstrates normal lung volumes with bilateral pleural effusions, left to greater extent than right with subjacent compressive atelectasis. b) Chest computed tomography of another child with JSLE: coronal reconstruction from contrast-enhanced CT demonstrates pericardial and pleural effusion a feature of serositis. The pulmonary trunk is dilated, a feature seen in pulmonary hypertension. There is also evidence of left axillary lymphadenopathy.
The C-reactive protein (CRP) is often high in contrast to the normal or minimally high levels observed in patients with other manifestations of active JSLE [18, 19].
Pleural effusions may also occur due to non-respiratory complications of JSLE, such as heart failure, or hypoalbuminaemia due to renal disease. Pulmonary infarcts or infection can cause haemorrhagic effusions associated with hypoxaemia [20].
The most common and severe acute life-threatening pulmonary complication of JSLE is acute lupus pneumonitis (ALP). It is rare, with a reported incidence of 1–4%, but with a mortality rate of 70–90% [14]. Children usually present with fever and respiratory distress. Inflammatory infiltrates in the interstitium and the alveolar wall cause diffuse alveolar damage, which is the hallmark of ALP [20]. Interestingly, ALP can be the initial presenting complaint of JSLE in half of the cases who develop this complication [14].
Although rare, alveolar haemorrhage (AH) is also an emergency in JSLE [21]. The child can present with shortness of breath, pyrexia, haemoptysis and a rapid decline in respiratory function. Any child with JSLE with respiratory symptoms and a drop in haemoglobin should be investigated for an AH. These children have high anti-dsDNA titres, and 90% of them have associated renal disease [3]. Other considerations for patients with JSLE and haemoptysis are infection and pulmonary embolism especially if there is underlying antiphospholipid antibody syndrome.
Thromboembolic disease is the most common complication of antiphospholipid antibody syndrome, and these children can experience dyspnoea, pleuritic chest pain and haemoptysis [22]. Risk factors include smoking, oestrogen-containing contraceptive pills and proteinuria from nephrotic disease.
In JSLE, subclinical lung disease is present in 60–70% of patients who undergo pulmonary function tests, for example restrictive lung disease or reduced DLCO [23, 24].
In JSLE, patients are prone to infections with common viral and bacterial organisms, as well as opportunistic fungal infections (including Pneumocystis jirovecii), and rarer viral agents such as cytomegalovirus. Several factors contribute to this increased risk including reduced pulmonary macrophage and phagocytic function, abnormal complement cell-mediated immunity, lymphopenia, immunomodulator medication, and even a lower airway clearance from muscle weakness. The lung interstitium is uncommonly affected in JSLE during the acute presentations, except in these pulmonary infections [20].
Any of these pulmonary complications in JSLE may occur independently or simultaneously, for example ALP and AH coexist with pleuritis [25]. They all carry a high mortality rate, with over half of the affected patients dying as a result [26].
Chronic pulmonary complications of JSLE
Chronic ILD is rare in JSLE, but it may occur after ALP [27]. These children present with a gradual onset chronic dry cough, pleuritic chest pain and exertional breathlessness. Anti-smooth muscle and anti-ribonucleoprotein antibodies are believed to increase the risk of ILD [25].
Chronic pulmonary hypertension in JSLE manifests as shortness of breath, with associated desaturation rapidly progressing to right-sided cardiac failure. Causes include chronic hypoxia in ILD, pulmonary vasculitis, chronic thromboembolism, and valvular cardiac involvement. Raynaud's phenomenon and the presence of antiphospholipid antibodies are risk factors for pulmonary hypertension in JSLE [28].
Shrinking lung syndrome in JSLE results in reduced lung volumes, with raised hemi-diaphragms and basal atelectasis seen on chest radiography [29]. A proposed explanation for this is the progressive diaphragmatic and intercostal muscle weakness.
Juvenile dermatomyositis
JDM or idiopathic inflammatory myopathy (IIM) is the most common inflammatory myopathy in children. It has an incidence of 0.1 per 100 000 children [30] with a peak incidence at 5–14 years old [31]. It is a multisystem disease that can present with proximal muscle weakness, skin rashes including Gottron papules and heliotrope rash, and raised skeletal muscle enzymes [32].
Primarily, it causes neuromuscular weakness which leads to pulmonary complications through hypoventilation and aspiration pneumonia (figure 2b); sequelae of dysphagia and gastro-oesophageal reflux [33]. All patients with JDM should be assessed for dyspnoea, dysphagia and dysphonia at every clinical review.

a) Chest radiograph of child with MCTD. This PA chest radiograph demonstrates retrocardiac left lower lobe consolidation; note increased retrocardiac opacity, indistinct bronchovascular markings and loss of cardiophrenic silhouette. Patent ductus arteriosus occluder device is in situ. b) Chest radiograph of a child with JDM with aspiration pneumonia. There is near total white out of the left hemithorax as a result of dense collapse consolidation of the left lung. There is wedge shaped consolidation in the right mid to lower zone.
As well as the complications related to weakness and/or dyscoordination, JDM-related ILD occurs in ∼10–20% of children. The most commonly described pathological findings in JDM-related ILD is nonspecific interstitial pneumonia and cryptogenic organising pneumonia [14]. While some of these children may be asymptomatic, others may experience a dry cough and worsening shortness of breath. JDM-related ILD bears a poor prognosis with 30% of affected patients dying within 5 years of diagnosis [3]. High serum levels of Krebs von den Lungen (KL-6), anti-melanoma differentiation-associated gene 5 (MDA5) antibodies, and interleukin (IL)-19 are associated with rapidly progressive ILD [34]. The presence of anti-Jo-1 antibodies is also a strong risk factor for developing this complication [35].
Juvenile systemic sclerosis
JSSc is typically characterised by symmetrical fibrous thickening and hardening of the skin combined with fibrous changes in internal organs such as the oesophagus, heart, kidneys and lungs [36].
The estimated incidence of JSSc ranges from 0.27 to 2.9 cases per million children per year [37]. The mean age of onset is 8–11 years old [38].
Severe lung disease is one of the major causes of death in children with JSSc. ILD is commonly found in JSSc, with rates of 65–90% in children. Nonspecific interstitial pneumonia is the most common form of ILD in JSSc [14].
Mixed connective tissue disease
MCTD was first described in 1972 as a CTD characterised by the presence of a specific autoantibody, now called anti-U1 ribonucleoprotein (RNP) [39]. It is often described as an “overlap syndrome” as it is associated with anti-U1 RNP antibodies and certain clinical features of JSLE, JSSc and JDM [40]. It often takes several years to diagnosis as the clinical features develop over time.
MCTD is rare in paediatrics, constituting up to 0.5% of the rheumatology workload [41].
Pulmonary complications occur in 75% adult patients with MCTD, and the proportion of affected children are believed to be comparable [3]. The complications include pleural effusions, pulmonary fibrosis and pulmonary hypertension, and are associated with a significant increase in morbidity and mortality [28].
Sjögren's syndrome
Sjögren's syndrome is a chronic multisystem condition characterised by a triad of xerostomia (dry mouth), xeropthalmia (dry eyes) and arthritis. It can be primary or secondary to conditions such as lupus [42].
The prevalence of ILD in Sjögren's syndrome varies from 9% to 75% of patients and is most commonly seen in secondary Sjögren's syndrome. Nonspecific interstitial pneumonia is the most common pattern of disease, but lymphocytic interstitial pneumonia is also seen [14].
Investigations
Pulmonary investigations in CTD have multiple roles including screening, diagnosis, and monitoring of disease progression and complications of treatment.
Blood tests
Acutely, raised inflammatory markers and white cell counts can indicate active inflammation, but also infection which is extremely important to exclude in these conditions.
A few specific autoantibodies (e.g. anti-RNP, anti-Jo-1) are associated with pulmonary disease in CTD, but their titres may not systematically correlate with disease activity [7].
Pulmonary function testing
PFT is useful for detecting subclinical disease, for assessing disease severity, and for long-term monitoring of respiratory complications. They include FVC, FEV1 and FEV1/FVC ratio, maximal inspiratory and expiratory pressures, as well as DLCO [43]. Abnormalities in PFTs may precede radiological changes. Regular outpatient lung function testing will detect pulmonary complications of CTD early, which will enable clinicians to promptly manage these conditions.
A restrictive spirometry (figure 3) is commonly seen in JSLE-associated ILD. An increasingly restrictive pattern may indicate its progression [44].

Pulmonary function testing in a patient with JSSc showing a restrictive pattern and reduced DLCO. VC: vital capacity; PEF: peak expiratory flow; FRC: functional residual capacity; TLC: total lung capacity; KCO: transfer coefficient of the lung for carbon monoxide; VA: alveolar volume; Hb: haemoglobin. For standardised residuals (SR): a value of >1.64 is the upper limit of normal, <−1.64 is below the lower limit of normal. Severity scale: mild = −1.64 to −2.5; moderate = −2.5 to −3.5; severe= <−3.5.
Children with pulmonary involvement in JDM may display restrictive or occasionally obstructive patterns; there is a restrictive pattern in the spirometry of 8% of patients with asymptomatic JDM [45].
In the clinic setting, DLCO is a sensitive indicator of pulmonary involvement, and impairment in DLCO can precede abnormal radiological findings. DLCO is decreased in ILD and may be low in almost half of patients with CTD, but this is not specific for ILD as it can happen secondary to pulmonary hypertension, which may also occur in CTD.
A recent study showed that 20% of patients with JSLE who underwent screening had a significantly reduced DLCO [46]. Acutely, an increase in DLCO can suggest AH, which is an acute complication of JSLE [47]. Shrinking lung syndrome in JSLE is characterised by a restrictive spirometry, and a normal or increased DLCO [29].
The frequency of screening and monitoring for pleuropulmonary complications in CTD remains controversial. Once lung disease is detected, spirometry with FVC, FEV1 and FEV1/FVC ratio should be undertaken every 3 months, and a full set of PFTs with DLCO can used longitudinally to monitor disease progression and remission every 6–12 months [7].
Assessment of oxygenation
Measuring oxygen saturation at rest and during exercise, as well as during sleep (through an overnight oximetry) helps to quantify the nature and the severity of the physiological impairment of gas exchange caused by the CTD-ILD [48]. The severity-of-illness score, described by Fan et al. [49], is determined at the initial assessment of children with ILDs and correlates with survival. A score of 1 to 5 is attributed as follows [49].
Score 1: Asymptomatic
Score 2: Symptomatic, normal room air oxygen saturation under all conditions
Score 3: Symptomatic, normal resting room air saturation, but abnormal saturation (<90%) with sleep or exercise
Score 4: Symptomatic, abnormal resting room air saturation (<90%)
Score 5: Symptomatic with pulmonary hypertension
Furthermore, the adequacy of oxygenation is being used as an outcome measure in current clinical trials, for example for the use of nintedanib in ILD [50].
Imaging
Plain film chest radiography may show consolidation or collapse, which is suggestive of infection, as exemplified in figure 2, or pleural lines which indicate pleural effusions in cases of pleuritis [28].
In the clinic setting, chest radiography has a low sensitivity and specificity for diagnosing ILD in CTD. In JSLE, chest radiography changes include reticular interstitial infiltrates or honeycomb changes, but PFTs with DLCO are twice as sensitive at picking up abnormalities compared with chest radiography [3].
Typical chest radiograph changes, such as ground-glass opacities and reticular patterns, are only seen in 10–25% of children with JSSc-related lung disease [3].
CT of the chest
A chest CT is more sensitive than a chest radiograph in the early detection of asymptomatic CTD-related ILD. However, performing CT for detecting pulmonary complications in children with CTD is a controversial area [7]. In very young children, a chest CT may necessitate a general anaesthesia with inherent risk for the child. There are also concerns about the dose of radiation from a CT scan for a child, but evidence shows that the newer low-dose CT parameters can give valuable information to guide diagnosis of pulmonary complications in CTD [51].
An array of techniques can be employed to maximise the potential quality of CT imaging and hence improve interpretation of the results. Prone position CT scanning reduces posterior dependent atelectasis. In younger children, controlled ventilation CT optimises full inflation [51]. In older children, full-inflation inspiratory CT and spirometer-controlled full-inflation CT can be employed [7].
Most of the data on imaging in CTD comes from adult studies, and shows that a CT detects pulmonary changes in between 8% and 70% of adults with JSLE, where the most frequent abnormality detected was ILD [3]. These studies suggest that a CT is more sensitive at diagnosing ILD in asymptomatic patients when compared with PFT with DLCO.
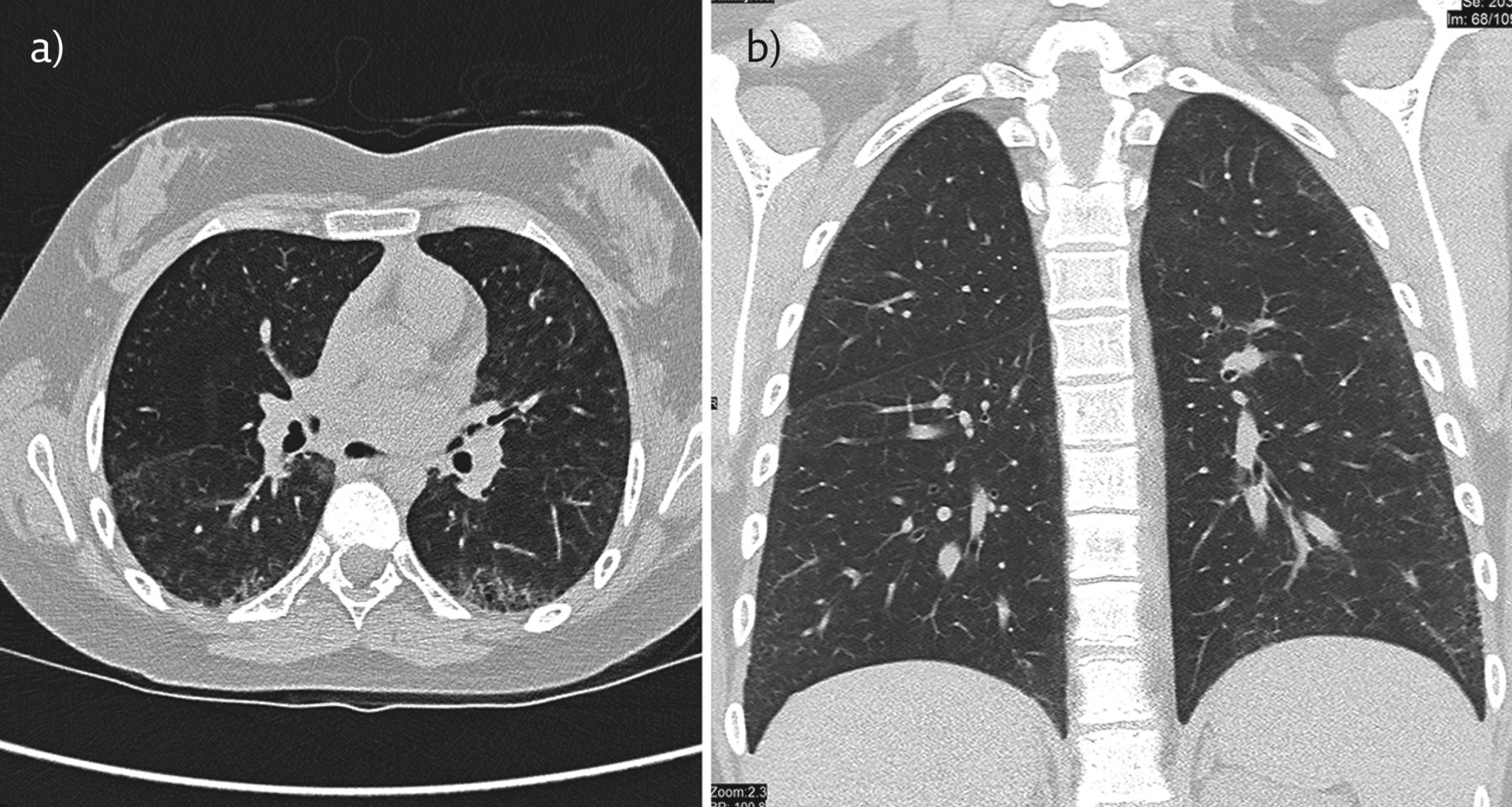
Findings of ILD on CT initially show an inflammatory state, which includes ground-glass opacities, reticular shadows and diffuse centrilobular nodularity. As ILD becomes more advanced, the CT changes denote a fibrotic state with honeycombing, bronchiectasis, and non-emphysematous cysts [52]. A typical chest CT of a teenager with JSSc is shown in figure 4. Staging of ILD on a CT scan can be classed as limited (involving <20% of the lung parenchyma), borderline (20–25% lung involvement), or advanced (>25% lung involvement) [53, 54].

Chest CT of a teenager with JSSc. a) Axial and b) coronal CT reconstructions: There is widespread intralobular septal thickening, predominating in the subpleural region. The apical segment of the left lower lobe demonstrates honeycombing indicating fibrosis. The fine subpleural ground-glass opacities in the right lower lobe indicates the presence of interstitial inflammation.
In JDM, a case–control study showed that 37% of patients were found to have abnormalities on chest CT including ILD and airways disease, but also chest wall calcinosis. In that cohort, only 26% of children had restrictive patterns on PFT, which shows that a CT may be more sensitive than PFT at detecting pulmonary complications [55].
CT imaging and Doppler ultrasounds are used to investigate for venous thromboembolic complications in children with JSLE acutely presenting with chest symptoms, especially in those having risk factors [22].
A chest CT may be a precursor investigation to guide a targeted BAL or a lung biopsy in CTD.
Not only is a chest CT diagnostic, it also aids the clinician in deciding on treatment options, guides the prognosis for the patient and helps in the monitoring of the response to treatment.
Bronchoscopy
In an acute setting, bronchoscopy allows the visualisation of the airway. An inflamed mucosa provides evidence of capillaritis, which occurs in AH in JSLE. BAL samples are usually bloodstained in AH. Equally the presence of haemosiderin-laden macrophages even in non-bloodstained BAL shows that there has been recent bleeding [20].
Thick secretions on bronchoscopy may provide evidence of pulmonary infections. The BAL aids in the identification of the organism. The culture and sensitivity results help guide the antimicrobial regime. The BAL cytology helps to delineate inflammatory cells and may hence help to exclude infectious processes, and subsequently point to a diagnosis of ILD. A lymphocytic or eosinophilic predominance in the BAL cell count can indicate drug-induced organising pneumonia [7].
The lipid-laden macrophage index in the BAL sampling can complement the workup for other associated conditions which lead to recurrent chest infections in CTD, such as aspiration and gastro-oesophageal reflux [56].
Thoracentesis
Needle aspiration of pleural effusions can be a useful diagnostic test. Pleural aspirates are exudative in pleuritis and empyema.
Lung biopsy
A lung biopsy may be indicated where the diagnosis is unclear, especially in the rarer childhood CTDs or in the presence of atypical features like hilar or mediastinal lymphadenopathy.
In JDM, due to the disease being so rare in children, a lung biopsy may still be needed to confirm the diagnosis of ILD, in addition to the high-resolution CT [28]. 50% of lung biopsies provide useful information to make a diagnosis of ILD in CTD. Typical ILD histopathology findings include desquamative interstitial pneumonia, nonspecific interstitial pneumonia and lymphocytic interstitial pneumonia [7].
A protocol for optimal lung biopsy outcome is detailed by the Children's Interstitial Lung Disease network [57]. The preferred approach is the use of a video-assisted thoracoscopic surgery, to obtain lung samples from various sites (a minimum of two) with the avoidance of the lingula and the tip of right middle lobe [52].
If a lung biopsy is needed, it is advisable to obtain it early in the course of pulmonary disease with the least amount of steroid use.
Echocardiography
It is recommended that all patients with JSLE undergo baseline echocardiography for early recognition of cardiac involvement, which can include pericarditis (commonest), myocarditis and non-infective endocarditis, and very rarely even pancarditis [58].
Pulmonary arterial hypertension (PAH) can be uncommonly associated with CTD [59, 60]. The clinical presentation of PAH can be nonspecific but can present with respiratory symptoms for e.g. tachypnoea, exercise intolerance, chest pain, syncope and progressive or refractory hypoxia [61]. Deterioration in CTD is usually rapid when associated with PAH and the presence of PAH in CTD attributes the highest score in severity-of-illness score [49].
If PAH is suspected, an echocardiography is performed. PAH is defined as a pressure >20 mmHg [59] and may complicate CTD. It can be suggested by an echocardiogram showing elevated right ventricular (RV) systolic pressures. The most precise echocardiographic method for measuring RV (and hence pulmonary artery) systolic pressure is to establish the velocity of the tricuspid regurgitant (TR) jet. The position of the interventricular septum at end systole is also a useful adjunct and offers a rough estimate of RV systolic pressure, but accurate quantification of pulmonary artery pressures is achieved only if a TR jet is measured [62].
Gastrointestinal assessment
Patients with CTD may suffer from gastro-oesophageal reflux disease and aspiration, comorbidities which can further compromise lung health. Investigations which may help in the diagnosis include a pH study, an impedance study, an upper gastrointestinal contrast study or a videofluoroscopy [63].
Treatment
General strategies
The treatment goals in children with CTD include controlling the underlying inflammatory condition and the prevention and/or treatment of pulmonary complications.
A multidisciplinary team approach including respiratory paediatricians, paediatric rheumatologists, radiologists and pathologists is necessary in children with CTD for optimal outcomes.
Supportive care is essential. This includes adequate nutritional support, avoidance of secondhand smoke and e-cigarette vapour exposure and other irritants, the use of prophylactic antibiotics, prevention of infection through vaccination (e.g. the yearly influenza vaccine) and making sure that children have mounted a good response to their routine childhood vaccinations.
Supervised rehabilitation programmes and the provision of family support are important for a holistic approach to the child's care. In cases of severe ILD leading to inadequacy of oxygenation at rest and/or during sleep, long-term oxygen therapy is considered [64].
Due to the heterogeneity of CTD and some unanswered questions regarding the underlying aetiologies and pathogenesis, the evidence-based pharmacological treatments for the pulmonary manifestations in each condition varies.
JSLE treatment grouped by complications
Infection
As infection is the leading cause of death in patients with JSLE [65], all pulmonary changes should be considered infectious until proven otherwise. Therefore, a thorough and aggressive approach to investigation is required including bronchoscopy and lung biopsy. If a proven pathogen is isolated, this should be managed appropriately with the correct antibiotic choice. In some instances, an empirical antibiotic regime may be used.
Pleuritis
Mild cases can be managed with oral analgesia and oral steroids. In moderate to severe cases, patients usually require oxygen, analgesia and intravenous methylprednisolone for 3 days, followed by a short course of oral steroids. Although rare, refractory pleuritis and pleural effusions in JSLE have been treated with talc poudrage [66] or tetracycline pleurodesis [67].
Acute pneumonitis
This generally has a good response to steroids. However, if non-responsive to steroids and if chronic inflammation continues and shrinking lung syndrome develops, another immunosuppressant may be required, depending on the CTD and previous treatments if any.
Acute pulmonary haemorrhage
This is a life-threatening event that often requires ventilation and high-dose intravenous methylprednisolone. This is a sign of poor disease control; therefore, escalation of treatment is required. Cyclophosphamide has shown significant improvement in symptoms and lung function [16]. Rituximab, a monoclonal antibody which works by removing B-cells, may be helpful in reducing the frequency of recurrences of acute pulmonary haemorrhage in JSLE [68, 69].
ILD
There is no consensus on the pharmacological treatment of ILD. This may be due to the rarity of the disease, the difficulty in obtaining histological confirmation of ILD, and the various types of ILD. ILD presents with varying degrees of histology from inflammation to fibrosis and a wide spectrum of clinical manifestations from minimal symptoms to respiratory failure [14]. This makes it even more challenging for clinicians to make decisions regarding treatment options, observational strategies, optimal timings for interventions and the appropriateness of pharmacological agents for treatment [14].
As inflammation plays an important role in many forms of ILD, steroids are used in ILD [14]. There is a lack of evidence on the steroid regime that should be used in children; however, intravenous methylprednisolone is associated with fewer side-effects [52]. Although not specific for paediatric CTD-associated ILD, a European protocol does provide guidance on treatment approaches in children. Steroid-sparing agents such as hydroxychloroquine, azathioprine, methotrexate, cyclophosphamide, mycophenolate mofetil (MMF) and biologics such as infliximab and rituximab have been used in ILD with varying success, but again with no clinical trials showing their efficacy in children [14, 70].
A paediatric clinical trial is currently being conducted on nintedanib [50], an intracellular inhibitor of tyrosine kinases, which has already proven to inhibit processes involved in the progression of lung fibrosis in adults [71].
Those children with advanced ILD may require long-term oxygen therapy. Lung transplantation is an option for those children who progress to end-stage lung disease.
The development of pulmonary hypertension in ILD is associated with a poor prognosis and poor survival. Treatment consists of immunosuppressive drugs, mainly steroids and cyclophosphamide, as well as a pulmonary vasodilator, such as bosentan, sildenafil and epoprostenol, and oxygen therapy [72, 73]. Early intervention is recommended [49].
Treatment of other CTDs and their pleuropulmonary complications
JDM
In 2012, a European initiative called Single Hub and Access point for paediatric Rheumatology in Europe (SHARE) reached a consensus on the management of JDM [74]. At the time of diagnosis or disease flare-up SHARE recommended intravenous methylprednisolone followed by oral steroids. This is in combination with a steroid-sparing agent. Methotrexate is the preferred choice and works well in maintaining remission at a dose of ∼15mg·m−2 once weekly as a subcutaneous injection.
Cyclophosphamide is recommended in severe JDM-associated ILD. If JDM appears refractory to treatment, there are multiple other treatment options including biologics like rituximab, changing to anti-tumour necrosis factor (TNF) (infliximab/adalimumab) or using combination therapy with high dose methotrexate, ciclosporin A and intravenous immunoglobulins [74].
JSSc
The 2009 European League against Rheumatism (EULAR) recommendations for the treatment of JSSc recommends that cyclophosphamide should be considered for treatment of JSSc-related ILD, in particular for patients with progressive disease. This recommendation is based on two high-quality randomised controlled trials [75]. There are no published studies in children to date; however, in clinical practice cyclophosphamide is used.
MMF is an alternative treatment choice. It may be better tolerated and is associated with less toxicity than cyclophosphamide [76].
MCTD
There is no specific treatment for MCTD. Those children with severe myositis and systemic involvement such as lung disease require high-dose steroids and cytotoxic drugs such as cyclophosphamide. Other drugs such as methotrexate, azathioprine, MMF and biological agents such as infliximab have been used [77]. Rituximab has also shown promising results [78].
Sjögren's syndrome
There is no clear consensus on treatment of ILD in Sjögren's syndrome. Steroids are often used. There is some evidence to suggest that azathioprine with and without steroids may be effective in treatment [79].
Iatrogenic complications of CTD treatment
The medications used in CTD are summarised in table 2.
Medications used in CTD
Medications used to treat CTD may have undesirable pulmonary side-effects. It is important for clinicians to be aware of this as ongoing use worsens the pulmonary complications. It may be hard to differentiate between natural progression of CTD-ILD, for which the medication has been started in the first place, and iatrogenic complications.
Methotrexate can lead to pulmonary toxicity and this has been reported mostly in adults. It may lead to hypersensitivity pneumonitis, which has an incidence of 1–2% [3], and patients experience shortness of breath, a dry cough and pyrexia. Interstitial or alveolar infiltrates may be seen on the chest radiograph. In addition, other complications such as reactive airways disease, pulmonary fibrosis, organising pneumonia and acute lung injury may be caused by methotrexate [20].
Biological DMARDs are a very effective treatment strategy for CTD, but can also cause ILD [80]. It is therefore crucial that any pre-existing ILD has been confirmed/excluded before commencing treatment with these agents.
Studies show that rituximab is associated with progression to ILD in adults. These patients often experience shortness of breath, pyrexia and tachypnoea with low oxygen saturation. The diagnosis is made by typical findings on a chest CT, along with a restrictive spirometry and decreased DLCO [81].
Other immunosuppressant drugs used in CTD can have pulmonary complications which are summarised in table 3 [3, 81, 82]. However, these are uncommon in children. In such cases, the offending drug must be stopped and the child may need high-dose corticosteroids.
Patterns of pulmonary injury caused by CTD medication
The use of immunosuppressant medication can favour opportunistic infections, which predispose these children to lower respiratory tract infections. Latent tuberculosis (TB) may be activated. Investigations to rule out latent TB are undertaken before starting any biological therapy [65]. In addition to opportunistic infections, infections with common pathogens can present atypically or could be difficult to treat in these individuals and this should be borne in mind when managing these children.
Conclusion
Clinical presentations of autoimmune CTD are variable and can be associated with many pulmonary complications, as discussed in this review.
Acute complications must be managed promptly, and clinicians need to be aware of the propensity of these children to infection. Outpatient reviews undertaken jointly by respiratory and rheumatology paediatricians enable the early detection of pulmonary complications in asymptomatic patients. Children are offered extensive lung function testing and are referred for imaging appropriately.
Treatment strategies include the use of immunosuppressive medication, aiming to halt the progression of pulmonary disease. This model of multispecialty combined reviews aims to improve the prognosis for patients with CTD.
Key points
Pleuropulmonary complications of CTDs are rare in the paediatric age group, but they are associated with high rates of morbidity and mortality.
Many children initially may not experience respiratory symptoms, and unless clinicians are vigilant with a high degree of clinical suspicion, these complications may go undetected.
Complications can be acute, potentially life-threatening (e.g. lower respiratory tract infection or alveolar haemorrhage), or chronic (e.g ILD and pulmonary hypertension) with significant morbidity.
Pulmonary complications should be monitored by assessment of clinical symptoms, performing pulmonary function testing and through the use of various imaging modalities on a regular basis.
Treatment includes steroids and other immunomodulators, including biological agents. To achieve the best possible outcome, affected children should be managed jointly by respiratory and rheumatology paediatricians.
Self-evaluation questions
1. Which of the following features on a chest CT is not typically found in CTD-ILD?
Ground-glass opacity
Reticular and reticulo-nodular pattern
Traction bronchiectasis
Atelectasis
2. Which of the following in a BAL would suggest alveolar haemorrhage as a complication of JSLE?
Elevated macrophage-laden lipid index
Presence of haemosiderin-laden macrophages
Cell cytology showing eosinophilia
Ziehl-Neelsen stain positive
3. Which of the following statements is incorrect regarding rituximab?
Rituximab is a monoclonal antibody that works by removing B-cells
Use of rituximab can lead to ILD
Live vaccines including influenza vaccine can be administered to patients receiving rituximab
Undertaking a Mantoux test is mandatory prior to starting rituximab
4. A child with ILD is seen in clinic. She has the skin features shown in figure 5. What is the most likely underlying diagnosis?
JSLE
MCTD
JDM
JSSc

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Skin rash in a child with ILD.
Suggested answers
1. d.
2. b.
3. c.
4. c.
Footnotes
Conflict of interest: None declared.
- Received August 11, 2020.
- Accepted September 1, 2020.
- Copyright ©ERS 2020
Breathe articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.