



Precision medicine in bronchiectasis
- Thomas Pembridge and
- James D. Chalmers⇑
- Division of Molecular and Clinical Medicine, University of Dundee, Ninewells Hospital and Medical School, Dundee, UK
- Corresponding author: James D. Chalmers (j.chalmers{at}dundee.ac.uk)
Abstract
Bronchiectasis, due to its highly heterogenous nature, requires an individualised approach to therapy. Patients experience symptoms and exacerbations driven by a combination of impaired mucociliary clearance, airway inflammation and airway infection. Treatment of bronchiectasis aims to enhance airway clearance and to address the underlying causes of inflammation and infection susceptibility. Bronchiectasis has multiple causes and so the pathophysiology leading to individual symptoms and exacerbations are different between individuals. Standardised investigations are recommended by international guidelines to identify the underlying causes of bronchiectasis. The process of identifying the underlying biology within an individual is called “endotyping” and is an emerging concept across chronic diseases. Endotypes that have a specific treatment are referred to as “treatable traits” and a treatable traits approach to managing patients with bronchiectasis in a holistic and evidence-based manner is the key to improved outcomes. Bronchiectasis is an area of intense research. Endotyping allows identification of subsets of patients to allow medicines to be tested differently in the future where trials, rather than trying to achieve a “one size fits all” solution, can test efficacy in subsets of patients where the treatment is most likely to be efficacious.
Abstract
Bronchiectasis, due to its highly heterogenous nature, requires an individualised approach to therapy. Treatment targets symptoms and exacerbations by aiming to improve mucociliary clearance and to reduce airway inflammation and airway infection. https://bit.ly/3ite4B2
Educational aims
To highlight the importance of personalising treatments of bronchiectasis on an individual basis to treat both underlying aetiology and treatable traits.
To understand which tests are required to identify underlying causes of bronchiectasis.
To describe how to best target specific treatments such as macrolides, inhaled antibiotics and mucoactive therapies to the patients with bronchiectasis most likely to benefit.
Introduction
Bronchiectasis is becoming increasingly common, and patients suffer significant morbidity and mortality with reduced quality of life [1]. The key features of the disease are chronic or recurrent infections, ciliary dysfunction, chronic inflammation, and lung tissue damage which interact to create a progressive disease process that has been referred to as a vicious “vortex” [2]. Bronchiectasis is a complex, heterogenous disease and its treatment is challenging [3]. International guidelines recommend a variety of treatments targeting each component of the vicious vortex [3]. Most treatments for bronchiectasis do not have a strong evidence base due to a lack of large randomised trials [3] and many trials in bronchiectasis have shown inconsistent results or have failed to reach their primary end-points [4]. It is thought this is because the disease is so heterogeneous, a one-size fits all approach to therapy is unlikely to be appropriate [5].
Personalised or precision medicine targets a patients’ individual pathophysiology to find the most effective course of treatment and, equally important, to avoid the use of ineffective therapies [6]. Groups of patients defined by a specific pathophysiological process or biomarker are referred to as endotypes [7]. The first use of the term endotype in the context of personalised medicine for respiratory disease is credited to Anderson [7], who reviewed the pathophysiology of asthma in 2008 and described endotypes as “a subtype of disease defined functionally or pathologically by a particular molecular mechanism or by a treatment response”. A particular clinical feature or biomarker that leads directly to a targeted treatment is referred to as a “treatable trait” [8]. Patients may have multiple treatable traits leading to multidimensional holistic care of patients. A personalised approach to the treatment of bronchiectasis tests patients for treatable traits and prescribes corresponding therapies [5].
This review discusses the optimal assessment of patients with bronchiectasis to identify treatable traits and the use of clinical assessment and biomarkers to optimise therapy.
Treating the underlying cause of bronchiectasis
Lonni et al. [9] identified the most common aetiologies of bronchiectasis as post-infective (20%), COPD (15%), connective tissue disease (10%), immunodeficiency (5.8%) and asthma (3.8%); while in a smaller study, Shoemark et al. [10] also found that primary ciliary dyskinesia (PCD) and allergic bronchopulmonary aspergillosis (ABPA) were also prevalent aetiologies of bronchiectasis at around 15% and 10%, respectively. All studies of the aetiology of bronchiectasis identify a high proportion (40–80%) of patients where no clear cause can be identified, referred to as idiopathic. Part of the reason for the high frequency of idiopathic bronchiectasis is a lack of standardised testing for underlying causes internationally. Nevertheless, despite systematic testing, a high proportion of idiopathic cases are still identified. This high prevalence of bronchiectasis of unknown aetiology demonstrates a clear need for continued basic research into bronchiectasis, to reveal more about the disease mechanisms of bronchiectasis as this will help to identify new aetiologies which can then be identified in patients and, ultimately, treated.
It is important to treat the underlying cause of disease to prevent further damage to the lung and prevent this from further driving bronchiectasis disease. Lonni et al. [9] found that aetiological testing led to a change in treatment in 13% of cases, while Shoemark et al. [10] found that treatment was affected by aetiology in 37% of cases. This is reflected by the prevalence of specific treatable aetiologies. The common aetiologies are summarised in table 1.
Common aetiologies with their clinical features, investigations and specific therapies
Treatable aetiologies
Immunodeficiencies, such as common variable immunodeficiency/agammaglobulinaemia, cause bronchiectasis due to recurrent lung infections as patients have low numbers of mature B-lymphocytes in the blood and lymph [11]. Immunodeficiency is treated using immunoglobulin replacement and prophylactic antibiotics to prevent infection. Although this is ineffective in preventing bronchiectasis in many cases, subcutaneous immunoglobulin was more effective than intravenous immunoglobulins at preventing bronchiectasis showing some benefit in preventing bronchiectasis is gained [12].
Nontuberculous mycobacteria (NTM) infections are treated using a combination of antibiotics over prolonged periods (often 18–24 months) and with varying success depending on the species present [13]. Mycobacterium avium and Mycobacterium abscessus are the most frequently identified species [13]. The decision over whether to treat NTM is challenging, since NTM can be a cause or a consequence of bronchiectasis and may be progressive or a transient infection that clears without antibiotic therapy.
ABPA is caused by hypersensitivity to Aspergillus fumigatus or other Aspergillus species [14]. Diagnosis is made by detection of elevated specific IgE to Aspergillus (e.g. >0.35 kUA·L−1) and total IgE, along with associated clinical features. Specific treatment is with corticosteroids with or without antifungal drugs.
Immunodeficiency, NTM and ABPA represent the most common treatable causes and are tested for routinely, but other important causes may be identified in specific cases. Cystic fibrosis (CF) can present in adults with bronchiectasis. A sweat test for CFTR function along with genetic testing to identify mutations in the CFTR gene may be considered, particularly in patients with childhood onset of disease, infection with Pseudomonas aeruginosa and Staphylococcus aureus, upper lobe predominant disease, or extrapulmonary features [15]. It is essential to identify CF as patients must receive multidisciplinary care within CF specialist clinics and may benefit from highly effective CFTR modulator therapies [16].
PCD causes bronchiectasis in 50% of affected cases by the age of 8 years and ubiquitously by adulthood [17]. PCD diagnosis alters the management of bronchiectasis by increasing the focus on airway clearance, consideration of cardiac abnormalities and infertility, and because patients experience earlier infection with P. aeruginosa and therefore require regular monitoring and initiation of inhaled antibiotics [18]. Testing for PCD is not performed routinely, but is considered in patients with onset of symptoms during childhood including neonatal respiratory problems, prominent upper airway/nasal symptoms, infertility or cardiac disease such as dextrocardia [19]. Alpha-1 antitrypsin deficiency is relatively uncommon in patients attending bronchiectasis clinics [20]. In some countries augmentation therapy is licensed. Some causes of bronchiectasis, such as connective tissue disease and inflammatory bowel disease, may be evident from the history and so do not require laboratory testing. Many of the underlying causes have suggestive clinical features as shown in table 1.
Due to the importance of treating these aetiologies, standardised testing should be carried out upon initial diagnosis to identify aetiology in all patients as this may alter management of the disease or reveal the need for treatments of the underlying cause. The European Respiratory Society (ERS) and British Thoracic Society, among others, provide guidelines for investigation of aetiology which include taking a medical history, measuring full blood count and total and specific IgE against Aspergillus, IgG, IgA, and IgM to identify any immunodeficiencies, with specific tests such as genetic testing for PCD or CF if the patient history suggests this is appropriate [3, 15]. The main causes of bronchiectasis and the associated treatments are summarised in table 1.
Even in those patients with an identified underlying cause, the disease is often identified at an advanced stage where treatment of the underlying cause is insufficient to reverse the disease, and therefore, patients require additional therapy. A prototype of this is advanced CF treated with CFTR modulators, which has been shown not to provide long term suppression of P. aeruginosa infection. Patients therefore require long term treatment for infection despite highly effective treatment of the underlying cause [21].
The vast majority of bronchiectasis cases worldwide are idiopathic and post-infective, and therefore, have no specific treatment. Within these groups there is still profound inflammatory, infective, and epithelial heterogeneity resulting in very different presentations and approaches. The next sections discuss how these different aspects can be evaluated and targeted.
Inflammation
Bronchiectasis is an inflammatory disorder and higher levels of inflammation have been found to correlate with poorer lung function and worse extent of disease [22].
In bronchiectasis, inflammation is typically driven by neutrophils, which is the most abundant inflammatory cell observed in sputum or bronchoalveolar lavage from bronchiectasis patients [23]. Degranulation of neutrophils or the formation of neutrophil extracellular traps (NETs) leads to the release of neutrophil elastase, which is a key mediator that impairs defence against infection and promotes lung function decline through degradation of elastin [23]. It is thought that serine proteases such as neutrophil elastase released in the inflammatory response also reduce mucociliary clearance, another factor in the vicious vortex of bronchiectasis. This could be through the cleavage of the CFTR channel protein, leading to mucus dehydration [24]. In addition, chronic inflammation from bronchiectasis reduces the number of cilia present on each epithelial cell, further reducing the capability to clear mucus. These effects demonstrate how each aspect of the bronchiectasis vicious vortex interact with one another to perpetuate the disease [2]. The dysregulated inflammatory response which is typical of bronchiectasis can also counterintuitively aid infection. Recent data shows that neutrophils in bronchiectasis undergo NETosis instead of killing bacteria through phagocytosis, particularly in severe bronchiectasis [23]. NETosis is intended as a mechanism to trap and kill bacteria, but key respiratory pathogens such as Haemophilus influenzae and P. aeruginosa are resistant to NET-mediated killing [23]. NETs may, therefore, be counterproductive by generating an environment that is more conducive to P. aeruginosa dominance.
However, neutrophils are not the only inflammatory endotype in bronchiectasis. Eosinophilic inflammation is increasingly recognised and can be identified using peripheral blood eosinophil counts, as has been described for COPD and asthma [25, 26]. To date, small studies suggest that around 20% of bronchiectasis patients, excluding those with underlying asthma, have eosinophilic inflammation [27–29]. Although the literature on the importance of eosinophils in bronchiectasis is limited, ABPA stands as a clear example of how Type 2 inflammation can contribute to both the development of bronchiectasis and exacerbations. Identification of eosinophilic inflammation is important as it requires a different treatment.
Current anti-inflammatory and immunomodulator treatments
Macrolide antibiotics have widely studied immunomodulator effects, which include effects on the epithelium, on neutrophils and neutrophil recruitment, and have recently been shown to reduce the formation of NETs [30]. An individual participant data meta-analysis of three randomised trials in bronchiectasis showed that macrolides over 6–12 months reduce exacerbation frequency by 51% as well as improving symptoms measured by the St George's Respiratory Questionnaire (mean 2.93 points) [30]. Intriguingly, among the groups of patients with the greatest response were patients infected with P. aeruginosa [30]. Macrolides have no in vitro activity against P. aeruginosa, and therefore, their efficacy in this patient population supports the idea that they work through an effect that is not directly antibacterial. International guidelines suggest considering macrolides for patients with three or more exacerbations per year [3]. Safety issues include gastrointestinal side-effects, hearing loss, cardiovascular effects, and the risk of inducing resistance in both conventional bacteria and NTM.
The most widely used anti-inflammatory medications globally are inhaled corticosteroids (ICS), which likely reflects extrapolation of their use from COPD and asthma. ICS are not recommended by international bronchiectasis guidelines and are, therefore, used inappropriately in many cases [3]. There are no large, randomised trials of ICS in bronchiectasis. In COPD it is now established that ICS are only effective in reducing exacerbations in a subgroup of patients with eosinophilic inflammation reflected by elevated blood eosinophil counts (e.g. >300 cells per μL) [31]. A recent post hoc analysis of a trial of two doses of fluticasone versus bronchodilators alone in bronchiectasis found improvements in quality of life only in patients with eosinophil counts in blood >3% [32]. This is early evidence that eosinophilic inflammation in bronchiectasis may also be responsive to ICS.
Future approaches
Numerous therapies which target inflammation are under development and may become available soon. Promising data showing the efficacy and safety of inhibiting dipeptidyl peptidase 1 (DPP1) has recently been published [33]. DPP1 activates neutrophil elastase, cathepsin G and proteinase-3 by cleaving the N-terminal dipeptide during neutrophil maturation [33]. By inhibiting DPP1, neutrophils are released from the bone marrow that lack active neutrophil elastase and other serine proteases and therefore inflammation is reduced. In the WILLOW phase 2 trial of DPP1 inhibition, two doses of brensocatib significantly reduced neutrophil elastase levels in sputum and this translated into prolongation of the time to first exacerbation compared with placebo [33]. The treatment was well tolerated. A large phase 3 trial is now underway (clinicaltrials.gov identifier: NCT04594369).
As noted earlier, alternative methods of targeting T-helper cell (Th)2 inflammation in bronchiectasis may represent a key treatment approach. In addition to corticosteroids, eosinophilic inflammatory endotypes of bronchiectasis may be treated with an anti-interleukin (IL)-5 or anti-IL-5 receptor monoclonal antibodies. A case series from Germany of patients with severe eosinophilic bronchiectasis showed marked improvements in exacerbation rate, quality of life and lung function in patients treated with mepolizumab or benralizumab [28]. A pilot study of mepolizumab in patients with bronchiectasis secondary to severe uncontrolled asthma showed a reduced number of eosinophils in both the blood and sputum, as well as increased forced expiratory volume in 1 s (FEV1) score and a significant reduction in the number of exacerbations per year [34]. These studies have small sample sizes and are limited by observational biases but should encourage formal trials. Thymic stromal lymphopoietin (TSLP) is an alarmin that activates allergic and non-allergic inflammatory responses including those mediated by Th2 inflammation, which in turn activates eosinophils. Encouraging data in asthma and the overlapping inflammatory processes in bronchiectasis and asthma suggest the potential for future trials in bronchiectasis [35].
Infection
Chronic infection is a central feature of bronchiectasis. Culture based studies suggest that globally the most common pathogens isolated by culture of sputum or bronchoalveolar lavage are P. aeruginosa and H. influenzae. Other frequently isolated organisms include Moraxella catarrhalis, S. aureus, Streptococcus pneumoniae and the order Enterobacterales, which includes organisms such as Escherichia coli and Klebsiella pneumoniae for example [36–39].
Patients with positive sputum cultures for pathogens have a higher frequency of mortality (11.8% over 4 years versus 6.0% for those not infected in one study) [40] and more frequent exacerbations. Once established, infections in bronchiectasis may become chronic and sputum cultures are persistently positive. This is particularly the case for P. aeruginosa, which forms robust biofilms and is highly resistant to antibiotics [41, 42]. Patients are at much higher risk of mortality in the presence of P. aeruginosa, estimated as three times higher than patients without P. aeruginosa infection in one meta-analysis, and also have higher exacerbation rates and hospitalisation rates [43]. NTM pulmonary disease is an important cause of bronchiectasis, with higher rates reported in Asia and the USA than in Europe [39, 44, 45]. It is important to identify NTM pulmonary disease as it has a specific treatment with prolonged antibiotic therapy. Recent data suggests that inhaled amikacin can be effective in refractory cases of M. avium infection [13, 46].
Culture remains the key tool available to clinicians for the diagnosis of infection, but molecular techniques, such as quantitative PCR and sequencing, are gradually emerging as part of clinical practice [47, 48]. Studies into the airway microbiome in bronchiectasis have predominantly used sequencing of amplicons of the 16 s rRNA subunit, to date, and have provided useful insights [49–53]. Specifically, sequencing has confirmed associations between higher relative abundance of organisms such as Haemophilus and Pseudomonas with exacerbations [54], while also confirming the relationship between Pseudomonas and increased mortality [55]. As conventional “pathogens” such as Pseudomonas and Haemophilus become dominant, less abundant “commensal” taxa become less frequent and this is reflected in microbiome studies through a reduction in diversity measured using diversity indices [49, 52, 53, 56]. Studies show relationships between reduced microbial diversity and lower FEV1, more frequent exacerbations and increased mortality [49, 52, 53, 56]. To date, studies have been unable to resolve the “chicken or egg” question of whether overgrowth of specific taxa such as Pseudomonas results in loss of diversity through interspecies competition, or whether antibiotic treatment over time contributes to reducing diversity and potentially creates a “potential space” for overgrowth of pathogens [51, 57, 58]. Supporting the idea that antibiotic treatment can cause long term changes in the microbiome, a study of long-term erythromycin showed increased relative abundance of Pseudomonas over 12 months [59].
Treatment of infection
How does this inform personalised treatment? First, regular sputum cultures are clearly useful in patients with bronchiectasis to monitor infecting bacteria, and to therefore guide antibiotic treatment at exacerbation [15, 60]. Regular sputum cultures may also identify the initial infection with P. aeruginosa or other less common organisms such as methicillin-resistant Staphylococcus aureus where eradication treatment may be offered [61–63]. There are no randomised studies demonstrating the effectiveness of eradicating P. aeruginosa but observational studies suggest clinical benefits. In a Dutch study of 60 patients with new P. aeruginosa infections, 36 (60%) were P. aeruginosa free for ≥1 year following therapy and 42% free of infection at 36 months [62]. In a study from Northern Ireland of 64 patients, 52% were P. aeruginosa free at 6 months and 36% at more than 1 year [63]. In both studies eradication consisted of systemic antibiotics with or without the addition of inhaled antibiotics. What is unknown is whether some of these patients would have spontaneously cleared P. aeruginosa without antibiotic therapy.
In patients with frequent exacerbations, it is clear that bacterial infection contributes to increasing exacerbation risk [23, 43, 64]. After optimising airway clearance and treating any underlying causes, such patients should be considered for prophylactic antibiotic treatment [3]. The most effective therapies in these circumstances are long-term macrolides, although whether these work primarily through an antibiotic or a non-antibiotic mechanism remains controversial [23, 30]. In patients for whom macrolides are not appropriate, including patients with suspected or confirmed NTM, prolonged QT interval or adverse effects, targeted antibiotic treatment can be considered [15]. There is little published data supporting the use of long-term amoxicillin or doxycycline for H. influenzae infection, but they are recommended as possible approaches by the British Thoracic Society guidelines and are supported by longstanding clinical experience [15, 65].
Inhaled antibiotics have potential advantages over systemic antibiotics by delivering broad spectrum antibiotics at high concentration directly to the site infection [66, 67]. ERS guidelines suggest use of inhaled antibiotics for patients with P. aeruginosa infection or as add on therapy for patients who continue to exacerbate despite oral antibiotic therapy [3]. Inconsistent results have been observed in randomised trials of inhaled antibiotics, and a review by Laska et al. [68] concluded a mean reduction in exacerbation frequency of 19% with a larger effect on severe exacerbations (reduced by 57%). The inconsistent results in trials most likely reflect patient selection, trial design and particularly the administration of antibiotics in a cyclical manner [69]. The latter is important as bacterial loads increase during off periods in patients receiving 28-day on/28-day off regimens (or equivalents such as 14-day on/off regimens), which allows increases in inflammation and worsening of symptoms which likely also increases risk of exacerbation [70–72].
How to identify patients that are more likely to respond to inhaled antibiotics? Recent data suggests that, in terms of symptoms (which are highly linked to exacerbations, since exacerbations are defined by a worsening of symptoms), inhaled antibiotics predominantly improve cough, sputum production and sputum colour [71]. Therefore, patients may be more likely to respond to therapy if these are their predominant symptoms. Only one biomarker has so far been identified for predicting inhaled antibiotic response. In the AIR-BX1 study and two studies of inhaled aztreonam, patients experienced marked improvements in symptoms if their baseline bacterial load was greater than 107 cfu·mL−1, a level of bacterial burden that is associated with more inflammation and increased exacerbation risk [73]. Patients did not experience an improvement with lower bacterial loads. Bacterial load measurement is not currently available as a routine test, but patients with higher bacterial loads would be expected to have greater sputum purulence, more persistently positive sputum cultures and more frequent exacerbations [71, 72].
Bronchospasm is experienced by 10–20% of patients with bronchiectasis receiving inhaled antibiotics, particularly inhaled aminoglycosides and aztreonam [68, 74, 75]. Consequently, as well as identifying the patients most likely to benefit it is important to identify the patients most likely to experience harm. Most trials exclude patients with low lung function (FEV1 <25% or 30%) and the study by Crichton et al. [71] identifies increased exacerbations in patients with prominent wheeze (75% increase) and absence of cough and sputum purulence. Therefore, it seems wise to avoid inhaled antibiotics in such patients. In patients with lower lung function it may be appropriate to consider colistin rather than aminoglycosides as trials, to date, suggest lower rates of bronchospasm with this agent [76]. It is advisable to conduct a supervised first dose of inhaled antibiotics with measurement of lung function post-procedure to monitor for the development of bronchospasm.
In the future it is tempting to speculate that biomarkers may help to identify the patients most likely to benefit from inhaled antibiotics, which may include bacterial load or markers of inflammation such as neutrophil elastase [22, 73, 77]. An area requiring greater exploration is the third of patients with bronchiectasis who do not have chronic infection by conventional definitions and who have microbiome profiles with “commensal” taxa such as Streptococcus, Veillonella, Prevotella, Rothia, Neisseria and others [51]. Do some of these taxa contribute to pathology and exacerbations despite not being traditionally reported on culture? Recent data suggesting that the balance or interactions between different organisms strongly contributes to exacerbation risk suggests that a more personalised approach to targeting antibiotic treatment (or avoiding antibiotic treatment in those where infection is not contributing to risk) may be possible in future [50].
Mucociliary clearance
Cilia are microscopic hair-like structures found on the airway epithelial cells, their role being to carry particulates trapped in mucus out of the lung to protect against infection [78]. Mucus clearance from the lower lung by cilia is therefore required to a maintain lung homeostasis. In bronchiectasis, the bronchioles widen which reduces the ability of cilia to remove mucus from the lung, causing mucus obstruction of the airways. This creates an ideal niche for pathogens and a “reservoir” of bacteria from which infection can spread to other areas of the lung [79, 80]. The resulting higher bacterial loads can then drive further neutrophil recruitment, which increases inflammation [70].
The severity of cilia disruption varies greatly between bronchiectasis patients, with some having somewhat normal cilia function while others’ cilia are totally immotile. Ciliary dysfunction can be primary, as in PCD, or secondary to inflammatory damage, or bacterial proteins from organisms such as P. aeruginosa which are directly ciliotoxic [81, 82].
Compared to inflammation and infection, mucociliary clearance is poorly studied in non-CF bronchiectasis. Effective mucus clearance is a function of ciliary beat frequency, stroke length and stroke pattern, and the characteristics, viscosity and volume of mucus [78]. Clinical tests for most of these functions do not exist and little is known about how they are regulated, although high-speed video-microscopy offers a chance for a trained expert to visualise the cilia beating to assess cilia impairment and is used in the diagnosis of PCD among other tests. The characteristics of mucus can be evaluated through direct measurement of mucins, mucus rheology and mucus weights (wet/dry). Ramsey et al. [83] recently published the most detailed analysis of mucus properties in bronchiectasis. They showed that, compared with controls, bronchiectasis patients had increased mucins and hyperconcentration of mucus. Importantly, the study showed that increased sputum solids were associated with disease severity and that mucus hyperconcentration could be altered by hypertonic saline.
Treatment of impaired mucociliary clearance
The key treatment approach in bronchiectasis is the performance of airway clearance exercises by patients, ideally demonstrated by and supported by a trained respiratory physiotherapist [3]. Although regular physiotherapy can improve mucus clearance and quality of life, many patients continue to have a high burden of mucus symptoms despite this. Devices such as positive pressure devices may also aid mucus clearance. Mucoactive drugs aim to modify the sputum to enhance mucus clearance. Hypertonic saline can be prescribed to create an osmotic pressure to increase the water content of mucus and reduce its viscosity. However, the efficacy of inhaled hypertonic saline in bronchiectasis is under question with a recent meta-analysis showing no greater effect of hypertonic saline than control (typically isotonic saline) [84]. Inhaled dry powder mannitol is a therapy with a similar mechanism of action to hypertonic saline, which increases mucus hydration and stimulates coughing. It failed to show a benefit, in terms of reduced exacerbation rates, in a phase 3 bronchiectasis trial [85]. A reanalysis of that trial recently showed that patients with high levels of baseline symptoms responded with a marked reduction in exacerbation rates [86]. This suggests that mucoactive drugs, perhaps unsurprisingly, are most likely to be beneficial in patients with prominent mucus symptoms. Other mucoactive therapies such as N-acetylcysteine or carbocisteine await testing in large scale trials but are widely used to reduce sputum viscosity.
Future approaches to mucociliary clearance
Sputum and cough remain the most important symptoms for patients with bronchiectasis [87], and therefore new therapies are needed to improve these features. It has been suggested that there is a subset of patients which have reduced CFTR function either due to heterozygous mutations or more importantly due to cleavage of the CFTR protein by serine proteases released by the immune system. Trials are now being performed to test whether CFTR modulation will be effective in bronchiectasis patients without CF (clinicaltrials.gov identifier: NCT04396366).
Additional considerations and treatable traits
Bronchiectasis patients commonly present multiple treatable traits that are not captured by the three classic areas of pathophysiology described above. Airflow obstruction is identified in more than 50% of bronchiectasis patients and when associated with significant breathlessness is treated with inhaled bronchodilators. Some patients with bronchiectasis without spirometric airflow obstruction also have air trapping or increased airway resistance and may benefit from bronchodilators [3, 88]. There is limited evidence, but based on clinical experience and the established safety profile of these treatments they are recommended by the ERS guidelines for patients with significant breathlessness, and not limited only to those patients with spirometric airflow obstruction. Coexisting asthma and COPD should be identified and treated [89]. Patients with significant breathlessness should also be encouraged to attend pulmonary rehabilitation and evidence suggests this is at least as effective in bronchiectasis as in COPD [90].
Patients with bronchiectasis have an average age of >60 years and on average have three other associated comorbidities at presentation [91]. Patients with comorbid conditions have worse quality of life and increased mortality. Identification of extrapulmonary treatable traits is key to improving patients’ quality of life. Common extrapulmonary treatable traits include depression and anxiety, obesity or low body mass index (BMI), gastro-oesophageal reflux, rhinosinusitis, and cardiovascular disease such as heart failure. These can be readily identified through examination and clinical history. It is not possible to discuss in detail all the possible coexisting conditions but table 2 summarises the most common pulmonary, extrapulmonary and aetiological treatable traits in bronchiectasis [5].
Treatable traits in bronchiectasis
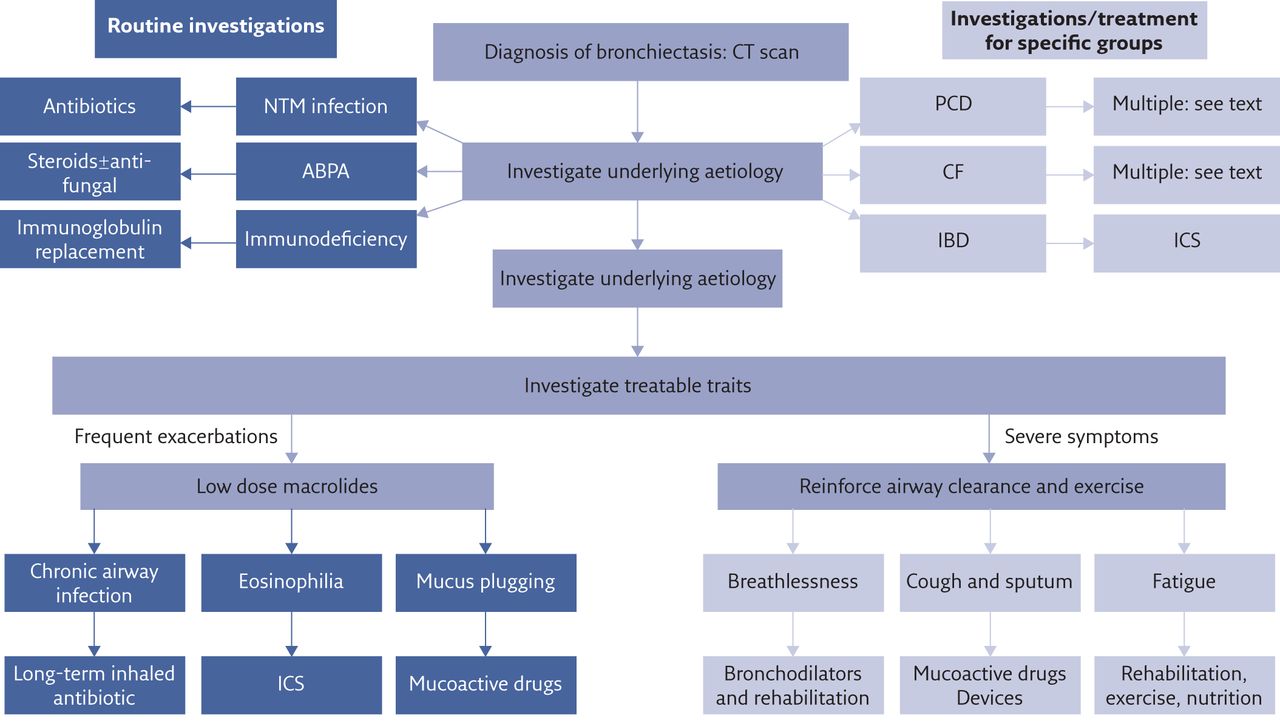
A schematic summarising the personalised treatment of pulmonary aspects of bronchiectasis is shown in figure 1.
{kind=link}
Schematic of personalised therapy for bronchiectasis. The underlying cause is investigated by standardised testing. All patients are tested for immunodeficiency, ABPA and NTM. Specific subsets of patients are tested for PCD and CF while inflammatory bowel disease (IBD) is typically apparent from clinical history rather than requiring testing. Airway clearance is recommended for all patients after which subsequent pharmacotherapy is dependent on the presence of treatable traits, targeted to reduce frequent exacerbations or reduce symptoms. It is acknowledged that patients will often have both exacerbations and symptoms and some therapies are able to improve both aspects.
Self-evaluation questions
1. A 27 -year-old man presents with a history of cough for over 10 years. It is productive of green sputum, and he has had two exacerbations in the past year. FEV1 is 72% predicted. On closer questioning, he had frequent chest infections and missed a lot of school during childhood. He had recurrent middle ear infections and has chronic rhinosinusitis for which he takes nasal corticosteroids. Sputum culture has grown P. aeruginosa. He has a normal BMI and no gastrointestinal symptoms. HRCT shows bilateral lower and middle lobe cylindrical bronchiectasis.
What is the most likely underlying diagnosis?
a) Post-infective bronchiectasis
b) PCD
c) ABPA
d) CF
e) NTM infection
2. A 69 -year-old lady presents with a chronic, persistent cough. It is productive of only a small amount of sputum daily, which is mucoid in colour. Her CT scan has been reported as showing bilateral lower lobe and middle lobe bronchiectasis. She has a BMI of 18.4 kg·m−2 and has lost an unspecified amount of weight in the previous 12 months. She has never smoked. She is breathless on walking 100 yards despite preserved spirometry with an FEV1 of 88% predicted. She has had three exacerbations in the past year requiring antibiotics, but does not feel that antibiotics are improving her symptoms.
What is the most likely underlying diagnosis?
a) ABPA
b) Sjögren's syndrome associated bronchiectasis
c) Idiopathic bronchiectasis with P. aeruginosa infection
d) Lung Cancer
e) NTM pulmonary disease
3. The same 27 -year-old gentleman from question 1, has had four sputum samples positive for P. aeruginosa in the past 2 years, which have failed to clear with repeated courses of oral ciprofloxacin and most recently a 2-week hospital stay for intravenous antibiotics. He feels better after antibiotic courses for around 4 weeks and then develops worsening cough, sputum production and malaise, and is experiencing repeated exacerbations. He has had 5–6 courses of antibiotics in the past year. He is practicing excellent chest physiotherapy having been trained by a professional physiotherapist. His sputum samples are negative for NTM, and he does not have ABPA. A repeat CT scan shows progressive bronchiectasis with extensive mucus plugging.
What is the next step in treatment (select all that apply)?
a) Start an inhaled corticosteroid
b) Start a long-term inhaled antibiotic
c) Start a long-term low dose macrolide
d) Start nebulised hypertonic saline
e) Attempt eradication of P. aeruginosa with systemic followed by 6–12 weeks nebulised antibiotics
4. A 56 -year-old lady has bilateral lower lobe bronchiectasis and presents with severe cough and extensive sputum production. She can fill a 50 mL sputum pot in an afternoon of coughing and finds this very distressing. She has had two courses of antibiotics in the past year and finds only a small reduction in her sputum production after she receives a course of antibiotics. She has not been diagnosed with asthma and has never smoked. She has a past history of GORD and hiatus hernia for which she takes a proton pump inhibitor and 10 years ago she had a colectomy for ulcerative colitis. She has sent multiple sputum samples which are negative for both typical bacteria and NTM. She is performing daily airway clearance exercises but finds the sputum comes up so frequently she does not need to do very much to expectorate. Laboratory tests including immunoglobulins, autoantibodies, ABPA serology and other tests are negative. Her blood eosinophil count is 280 cells per μL.
What is the next step in management?
a) Start an ICS
b) Start a long-term inhaled antibiotic
c) Start a long-term low-dose macrolide
d) Start nebulised hypertonic saline
e) Refer for consideration of a fundoplication
Conclusions
In conclusion, bronchiectasis is heterogeneous and optimal management requires a targeted approach. Inflammation, infection, and mucociliary clearance each become dysregulated and interact to create a self-sustaining vicious vortex. Therefore, effective treatments of each distinct component of the pathophysiology may be required to disrupt the vortex and achieve the best possible results.
Key points
Bronchiectasis has a variety of aetiologies some of which are essential to identify as they require a specific treatment.
Targets for treatment of bronchiectasis fall under three main categories: improving mucociliary clearance, treating and preventing infection, and reducing inflammation.
Bronchiectasis is a heterogeneous disease and therefore patients present with different types and severity of mucociliary dysfunction, infection and different patterns of inflammation. Personalised therapy is therefore essential.
Clinical tests such a history, symptoms, radiology, sputum cultures and blood counts can help to guide appropriate treatment to the right patient.
Suggested answers
1. b, PCD. This is a classic presentation of an adult with PCD. The history of recurrent otitis media and rhinosinusitis suggests ciliary dysfunction. The clinical suspicion of CF is less likely because there are no extrapulmonary features and the bronchiectasis is in the lower and middle lobes which is more suggestive of PCD.
2. e, NTM pulmonary disease. This patient has a typical clinical presentation of NTM pulmonary disease, which occurs most frequently in elderly females, often with low BMI and disease is most frequent in the middle lobe/lingula and lower lobes. Mycobacterium avium is the most common organism presenting in this way. Severe symptoms, fatigue, cough which is often dry, and poor response to conventional antibiotics are all common features.
3. c, but b and d may also be considered reasonable and may be required. This is a challenging question. The optimal treatment of P. aeruginosa infection in bronchiectasis is a subject of considerable debate and the evidence base is limited. In this case, treatment of the underlying causes and optimising airway clearance have been done. The next step would be to attempt to reduce exacerbation frequency. Macrolides have been shown to reduce exacerbations by more than 50% and are even more effective in patients with P. aeruginosa infection. Inhaled antibiotics are an alternative and may be favoured in patients with contraindications to macrolide. The efficacy data for inhaled antibiotics in bronchiectasis is currently less convincing than that for macrolides, but may clinicians still favour inhaled antibiotics based on clinical experience in CF.
Hypertonic saline may have a role here, given the extensive mucus plugging, but there is less evidence it would be effective in this case than a macrolide or an inhaled antibiotic. In reality it is not uncommon for patients such as this to receive all three of a macrolide, an inhaled antibiotic and a mucoactive drug.
ICS are unlikely to be beneficial in this case as the case provides no evidence of a responsive endophenotype. Eradication treatment is unlikely to be successful in a patient who has had multiple positive sputum samples over several years despite multiple courses of systemic antibiotics, although a small proportion of patients who receive inhaled antibiotics for the first time do achieve sustained culture conversion.
4. a, start an ICS. ICS are not typically recommended in patients with bronchiectasis but there are exceptions for a small number of treatable traits. First, patients with asthma and COPD may have indications for ICS independent of their bronchiectasis diagnosis and guidelines suggest following guidelines for asthma/COPD in this situation. Two other groups of patients may respond to ICS in bronchiectasis. The most well know is patients with IBD. IBD-related bronchiectasis is typically different in presentation and pathophysiology to typical bronchiectasis. Patients, as in this case, present with sterile inflammatory bronchorrhoea which responds to inhaled steroids. In this case the patient's sputum production improved within weeks and they were virtually asymptomatic at 6 months on high-dose ICS. There is emerging data that eosinophil counts >300 cells may also identify patients with bronchiectasis who benefit from ICS and this data is discussed in the article.
Footnotes
Conflict of interest: T. Pembridge has nothing to disclose.
Conflict of interest: J.D. Chalmers reports grants and personal fees from AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Insmed and Novartis, personal fees from Chiesi, Janssen, Grifols and Zambon, and grants from Gilead Sciences, outside the submitted work.
- Received August 7, 2021.
- Accepted September 29, 2021.
- Copyright ©ERS 2021
Breathe articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.